


ISSN: 0973-7510
E-ISSN: 2581-690X
Living organisms are naturally bestowed with unique and imitable qualities for maintaining ecological balance and earthworms are no exceptions. These so-called keystone species of terrestrial ecosystems are equipped with wonderful machinery, allowing them to nurture soil beautifully. Earthworm gut represents a potential microbial reservoir, having a complex interdependence with the host. The study aimed to profile bacterial community structure of three earthworm species belonging to two different life forms; Perionyx excavatus and Eudrilus eugeniae (epigeic), Polypheretima elongata (endogeic) respectively. Diversity analysis using 16S amplicon sequencing revealed that the dominant phyla were Proteobacteria (34.17-77.88) followed by Actinobacteria (13.43-35.54%), Firmicutes (1.69-15.45%) and Bacteroidetes (0.51-8.12%). The alpha diversity indices explicit similar gut microbiota of Perionyx excavatus and Eudrilus eugeniae and while higher alpha diversity was recorded in comparison to Polypheretima elongata gut. The taxonomic to the phenotypic annotation of 16S rRNA metagenomes revealed that dominance of Gram-negative bacterial community in all earthworm species while, Polypheretima elongata comprises higher percentage (78%) of Gram-negative bacterial community to Perionyx excavatus (32.3%) and Eudrilus eugeniae (38.3%). The oxygen requirement phenotypic analysis showed that all earthworm species were abundant with aerobic followed by anaerobic bacterial groups. Furthermore, functional metabolism phenotypic analysis revealed that a high abundance of ammonia oxidizers (29.3-80.2%), the gut microbiomes showed the relative abundance of sulphate reducer (22.6-78.7%), nitrite reducer (19.8-73.2%), dehalogenators (12.6-25.1%), illustrating in the role of these microbial communities in various degradation and bioremediation processes. The present study signifies the intrinsic gut microbiota of earthworm species for intensified biodegradation.
Metagenomic study, earthworm gut microbiota, ammonia oxidiser, bioremediation, biodegradation
Earthworms are the most dominant members of terrestrial ecosystems, play vital role in biogeochemical and nutritional rhythm of soil1,2. These unsung hero of the soil ecosystems alter the soil texture, regulate water content, maintain the availability of nutrients for the plants3 and regulate diverse biological functions by mixing the organic matter and other minerals within their gut4,5. In addition to shaping the soil structure, biogeochemical cycling and soil organic matter dynamics, earthworms have impacts on microbial communities in their gut, casts and drilosphere6. While the differences in the assimilation and digestion processes of earthworm indicate the possible occurrence of ecological group-specific gut microbial communities7. Thus earthworms also have an impact on the stability and the microbial diversity and properties of soil ecosystems8. Earthworm gut acts as a bioreactor and furnish favourable abode to microbes in comparison to the adjoining environment9. Earlier assumptions suggest that the earthworm gut microbiome depends on the bacterial diversity present in the surrounding environment1,10 and therefore the environment have a significant role in shaping the gut microbiomes1,11,12. In addition earthworms have been identified as epigeic, anecic and endogeic forms, depending upon their burrowing and food habits within various soil horizons13 which consequently regulating the core as well other bacterial communities within these three forms of earthworms. Moreover, the earthworm gut has the tendency to differentiate between the harmful and beneficial microbiota10,14. The earthworm harbours diverse microbial communities involved in metabolism, thereby maintaining the availability of essential nutrients and providing protection against pathogens1,10,14. The study evaluated taxonomic and functional profiling of gut microbiome of earthworms of diverse life forms and habitat.
Collection of samples
The earthworms used in the present study were collected from Nauradehi wildlife sanctuary (23°32’55.3″N 79°12’03.5″E; 600 m sea level) Sagar, Madhya Pradesh, India. Fifteen adult earthworms’ samples (five replicates for each experimental species) were collected following protocol described by Julka15. After considering morpho-anatomical parameters, worms were transferred into sterile plastic bags and delivered to Earthworm Biology Lab (23°50’03.7″N 78°47’01.2″E), Dr. Harisingh Gour Vishwavidyalaya (23°50’03.7″N 78°47’01.2″E), Sagar, Madhya Pradesh, India for further analysis.
Molecular identification of earthworms’ species
One adult earthworm was randomly chosen from each sterile polythene bag and characterized using mitochondrial molecular marker COI following standard protocol16. Table 1 depicts the accession number of studied samples. All the COI sequences of the present study are accessible on BOLD web portal under the research project ‘Diversity studies in earthworms of India’ (IEW). In addition, 22 COI sequences including outgroup were retrieved from NCBI and BOLD database for the molecular analysis.
Table (1):
Lists of samples, earthworms and outgroup taxa with accession numbers of COI gene sequence.
Sl. No. |
Species |
Gene |
Accession number (NCBI/BOLD) |
|---|---|---|---|
1 |
Drawida remiensis |
COI |
BOLD:ADH0513 |
2 |
Drawida remiensis |
COI |
BOLD:ADH0513 |
3 |
Drawida remiensis |
COI |
BOLD:ADH0513 |
4 |
Drawida remiensis |
COI |
BOLD:ADH0513 |
5 |
Eudrilus eugeniae |
COI |
MN125034.1 |
6 |
Eudrilus eugeniae |
COI |
MT410736.1 |
7 |
Eudrilus eugeniae |
COI |
KC122194.1 |
8 |
Eudrilus eugeniae |
COI |
KX832072.1 |
9 |
S2 |
COI |
BOLD:ACQ6907 |
10 |
Eutyphoeus Kempi |
COI |
BOLD:ADH6760 |
11 |
Eutyphoeus Kempi |
COI |
BOLD:ADH6760 |
12 |
Eutyphoeus Kempi |
COI |
BOLD:AAF0619 |
13 |
Eutyphoeus Kempi |
COI |
BOLD:AAF0619 |
14 |
Moniligaster aiyeri |
COI |
BOLD:ADH1655 |
15 |
Moniligaster aiyeri |
COI |
BOLD:ADH1655 |
16 |
Perionyx excavatus |
COI |
BOLD:ADC0803 |
17 |
Perionyx excavatus |
COI |
BOLD:ADC0803 |
18 |
Perionyx excavatus |
COI |
BOLD:ADC0803 |
19 |
S1 |
COI |
BOLD:ADC0803 |
20 |
Polypheretima elongata |
COI |
BOLD:AAF0305 |
21 |
Polypheretima elongata |
COI |
BOLD:AAF0305 |
22 |
Polypheretima elongata |
COI |
BOLD:AAF0305 |
23 |
Polypheretima elongata |
COI |
BOLD:AAF0305 |
24 |
S3 |
COI |
BOLD:AAF0305 |
25 |
Calomera littoralis (Out group) |
COI |
KX832072.1 |
Sequence alignment and data analysis for earthworm species identification
25 COI sequences were analysed on MEGA software using the Kimura two-parameter17. COI dataset was searched on NCBI, the Basic Local Alignment Search Tool (BLAST)
(http://blast.ncbi.nlm.nih.gov) to blast three query COI sequences and were aligned using Multiple sequence alignment program MUSCLE v3.8.31 (Multiple Sequence Comparison by Log-Expectation)18. Their phylogenetic estimation were inferred using neighbour-joining tree method following 1000 bootstraps using Molecular Evolutionary Genetics Analysis (MEGA X)19.
DNA extraction of gut microbiome
The same earthworm from each group was selected for gut metagenomic study, that tissue used for molecular identification of earthworms species. The worms were washed three time with distilled water and placed on separate sterile petri dishes (one per dish), after the couple of minutes worms were dissected to take out their gut. The metagenomic DNA was extracted using Qiagen Blood and Tissue Kit (Qiagen, USA) following provided protocol. Their quality was assessed on the 0.85% agarose gel and quantified on NanoDrop 2000 spectrophotometer (Thermo Fisher Scientific, USA).
16S rRNA gene amplification
The V3-V4 (469 bp) hypervariable region of 16S rRNA gene was targeted for amplification using primers 341F (5′-CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′)20. In order to differentiate all samples, the reverse primer was labelled with a specific barcode for each sample. For PCR reaction, total of 20 µl reaction mixture was prepared comprising 17 μl reaction buffer, AccuPrime Supermix, 1 μl of DNA template and 1 μl of each forward and reverse primer. The PCR Thermocycler was set at 95 °C for 5 min as initial denaturation step, followed by 35 cycles of 94 °C for 30s, 52 °C for 40s and 72 °C for 60 s, with a final extension step at 72 °C for 5 min. The same parameters were used for negative control of PCR product. Their integrity were analysed after mixing equal volume of 1X loading buffer ran on agarose gel electrophoresis. Subsequently, the intact and sharp bands between 400–500 bp were used for construction of library.
Construction of library and sequencing
The Qiagen Gel Extraction Kit was used to purify the PCR products and the library prepared using TruSeq DNA PCR-Free Sample Preparation Kit following protocol provided by manufacturer. The quality of library products were evaluated on Qubit 2.0 Fluorometer and Agilent Bioanalyzer 2100 system. The sequencing of constructed library was performed on an Illumina HiSeq 2500 platform using standard protocol21 at Nucleome Informatics Pvt. Ltd.
Data processing and In silico analysis
Paired-end raw sequences were filtered using FastQC22 on CosmosID’s bioinformatics pipeline (https://app.cosmosid.com; Rockville, MD, USA). Where, raw sequence files were uploaded to the CosmosID cloud application without set parameters or modified parameters. As reported earlier the application uses high-performance k-mer based algorithms and curated taxonomy databases (GenBook®) enable via the cloud interface23-28. Using CosmosID bioinformatics pipeline software the taxonomic community profiling, alpha diversity analysis (Chao1, Simpson and Shannon), Hierarchical clustering heatmap analysis at phylum and genus level was evaluated and plotted, to reveal microbial community composition in earthworms’ gut.
Taxonomic to phenotypic analysis
The taxonomic abundance table generated by CosmosID
(https://app.cosmosid.com) was uploaded on METAGENassist (http://www.metagenassist.ca/)29 for taxonomic to phenotypic profiling. The generated data were normalised following Paul et al30. Further analysis of phenotypic subsets, Gram stating oxygen requirement and metabolism having various phenotypic characteristics were corelated with given taxa, pie charts and bar graphs were plotted to depict the fraction of percent of taxa characteristic. The supervised pie chart and bar graph were employed for each metabolic phenotype analysis29.
Molecular identification of the species using the COI gene
Based on the molecular identification methods, these specimens were identified with the help of cytochrome oxidase subunit 1 (COI) gene partial sequence. The phylogenetic position of three query COI sequences was based on the BLASTN homology against the nucleotide sequence collection of the NCBI GenBank and BOLD sequence database and were identified as Perionyx excavatus (S1), Eudrilus eugeniae (S2), and Polypheretima elongata (S3). Obtained sequences showed 99% similarity with the available sequence in NCBI GenBank and BOLD databases were distantly related to the outgroup Calomera littoralis (Fig. 1).
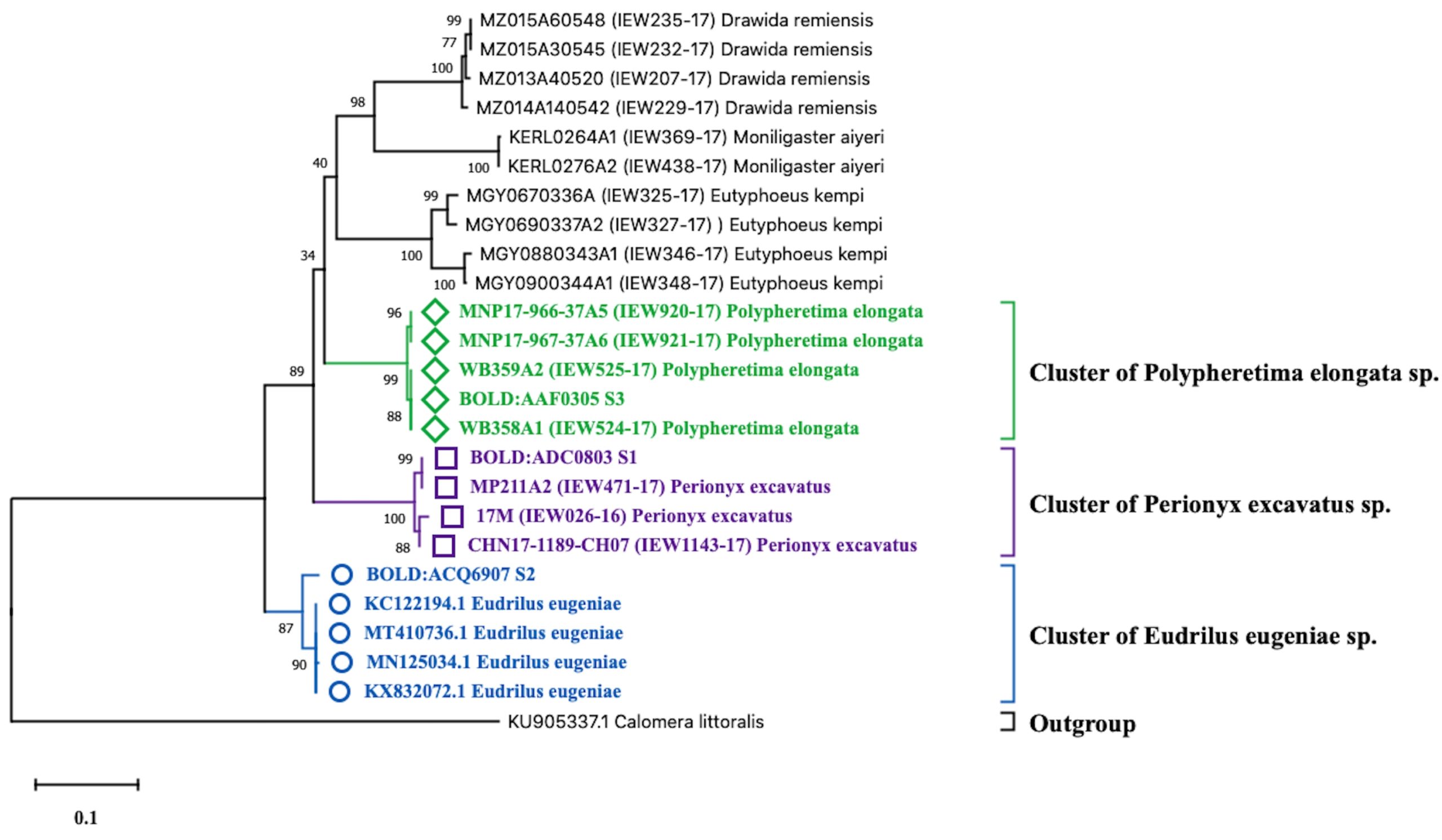
Fig. 1. Neighbour-joining tree with 1000 replication for selected strain of earthworms showing the relationship of S1, S2, and S3 with twenty four earthworm sequences and one outgroups (Calomera littoralis) using partial nucleotide sequence of COI gene.
Illumina Hiseq amplicon sequencing
Sequencing of the V3-V4 hypervariable region of 16S rRNA gene produced 2,54,807 high-quality reads of samples. The obtained reads were clustered into 1298 Operational Taxonomic Units (OTUs). The maximum, minimum and average number of reads per sample were 91938, 77455 and 84935 respectively.
Bacterial composition in earthworms’ gut
99.98% reads hit with k-mer markers were accounted for bacterial sequences. The relative abundances of microbial communities across the earthworms species gut were analysed at the phylum and genus levels. Ten bacterial phyla were detected in all samples. As shown in Figure 2 the majority of reads belonged to Proteobacteria, Actinobacteria, Firmicutes, Bacteroidetes and Chloroflexi, which accounted for 90-95% of total classified sequences (Fig. 2) which represents “core microbiota” of earthworms gut. While, multiple specific fluctuations were recorded in the proportions to relative abundance of various taxa from phylum level to genus level. Bacterial phyla Actinobacteria (35.54%), Proteobacteria (34.17%), Firmicutes (8.98%), Bacteroidetes (8.12%) and Chloroflexi (3.05%) were the most abundant in Eudrilus eugeniae, comprising 90% of the total microbiota. Corresponding the above, Proteobacteria (36.10%), Actinobacteria (31.64%), Firmicutes (15.45%), Bacteroidetes (4.01%) and Chloroflexi (4.86%) were abundant in Perionyx excavatus sp. gut occupied 93% of the total microbiome. While, Polypheretima elongata sp. gut inhabited Proteobacteria (77.88%), Actinobacteria (13.43%), Firmicutes (1.69%), Chloroflexi (0.65%) and Bacteroidetes (0.51%) carrying 94% of total microbiota (Fig. 2). The sunburst chart depicted the relative abundance of dominant genus in Polypheretima elongata gut, were Aeromonas (47.72%) followed by Enterobacter (7.44%), and Citrobacter (2.43%) (Fig 3A). While, Demequina (5.37%) followed by Mesorhizobium (4.27%), Cellulomonas (2.62%) and Rhodoplanes (2.47%) were dominant in Perionyx excavatus gut (Fig 3B). And, Demequina (7.86%) followed by Flavobacterium (2.81%), Salinibacterium (2.20%), Mizorhizobium (1.74%) and Cellulomonas (1.50%) were more abundant in Eudrilus eugeniae gut (Fig. 3C).
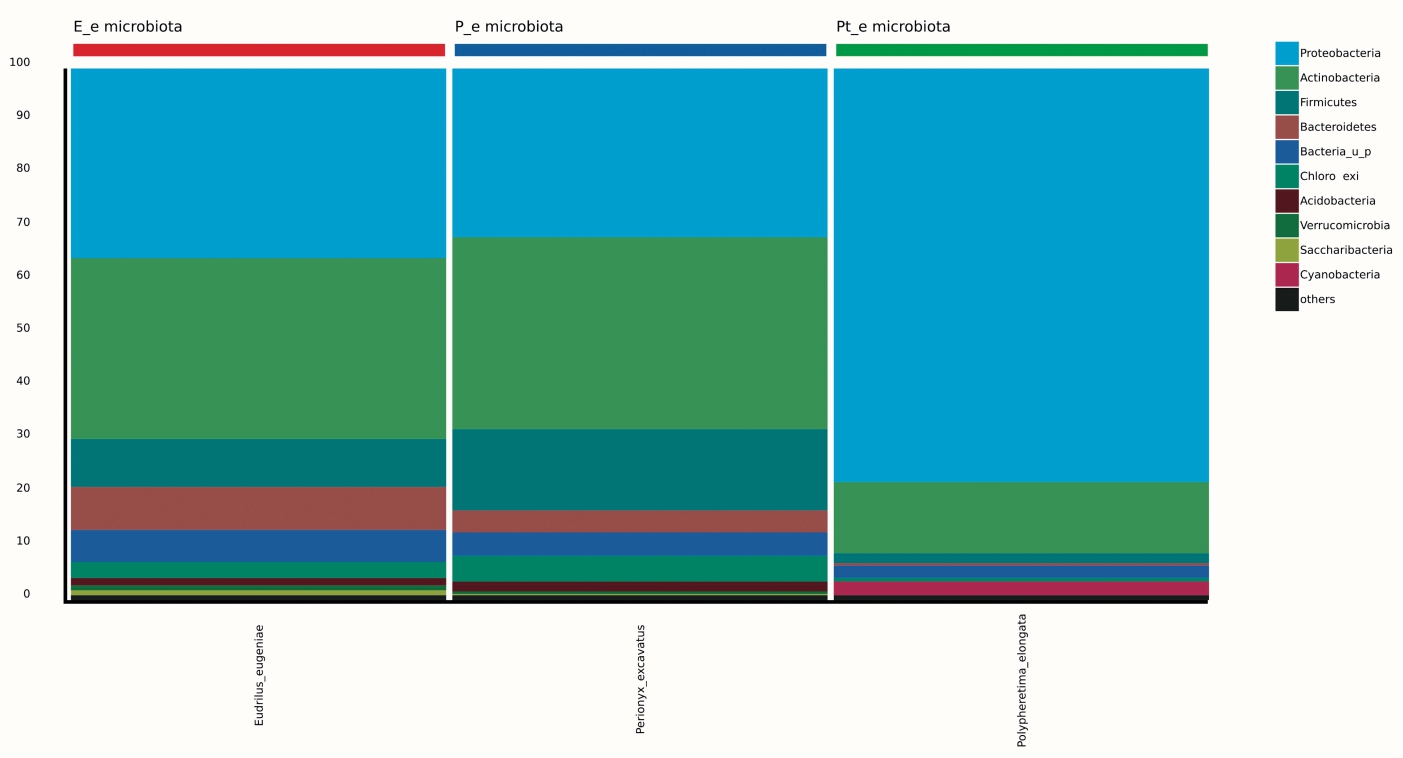
Fig. 2. Phylum level relative abundance of bacterial communities in different species of earthworms viz Polypheretima elongata; Perionyx excavatus; and Eudrilus eugeniae.
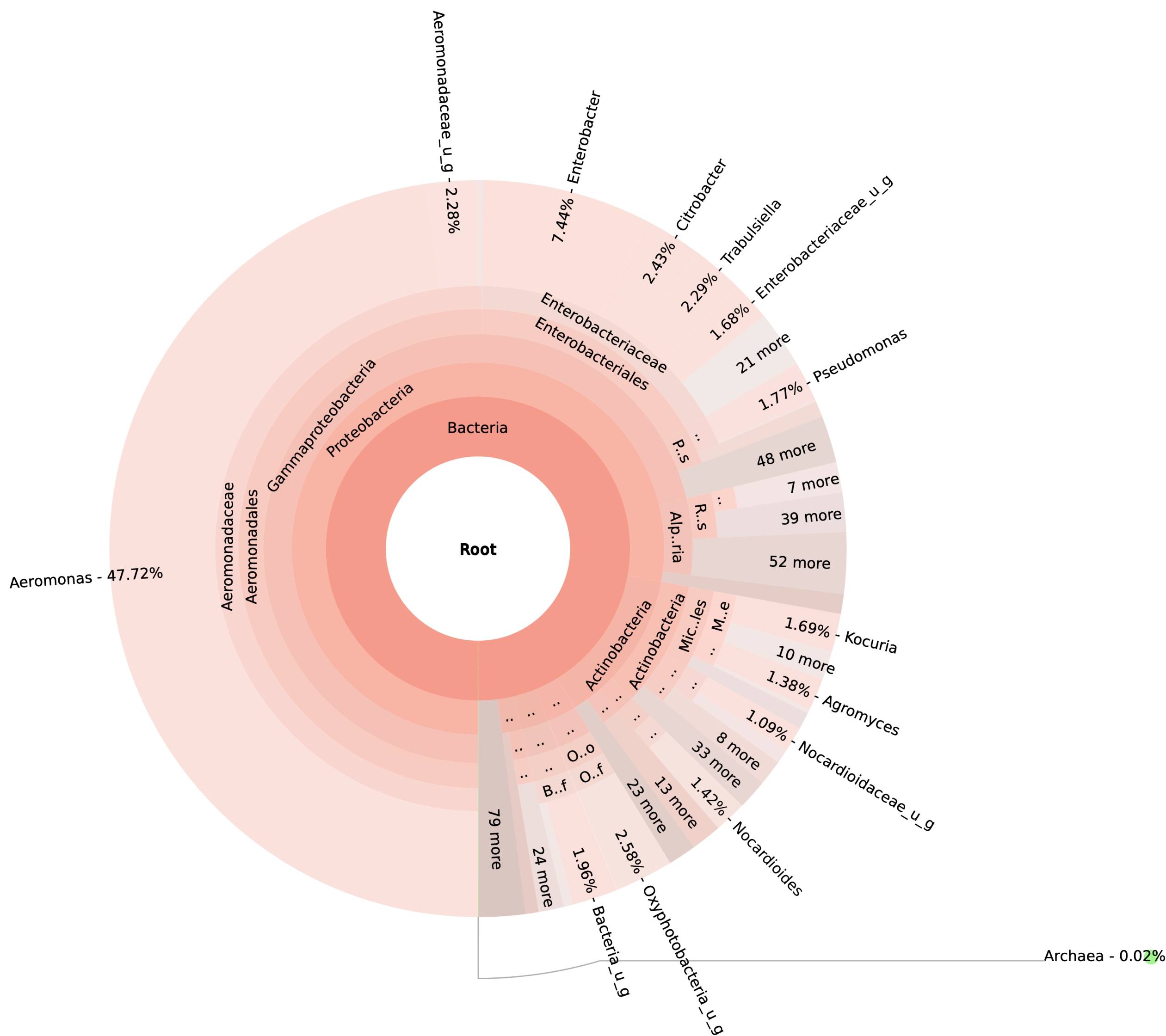
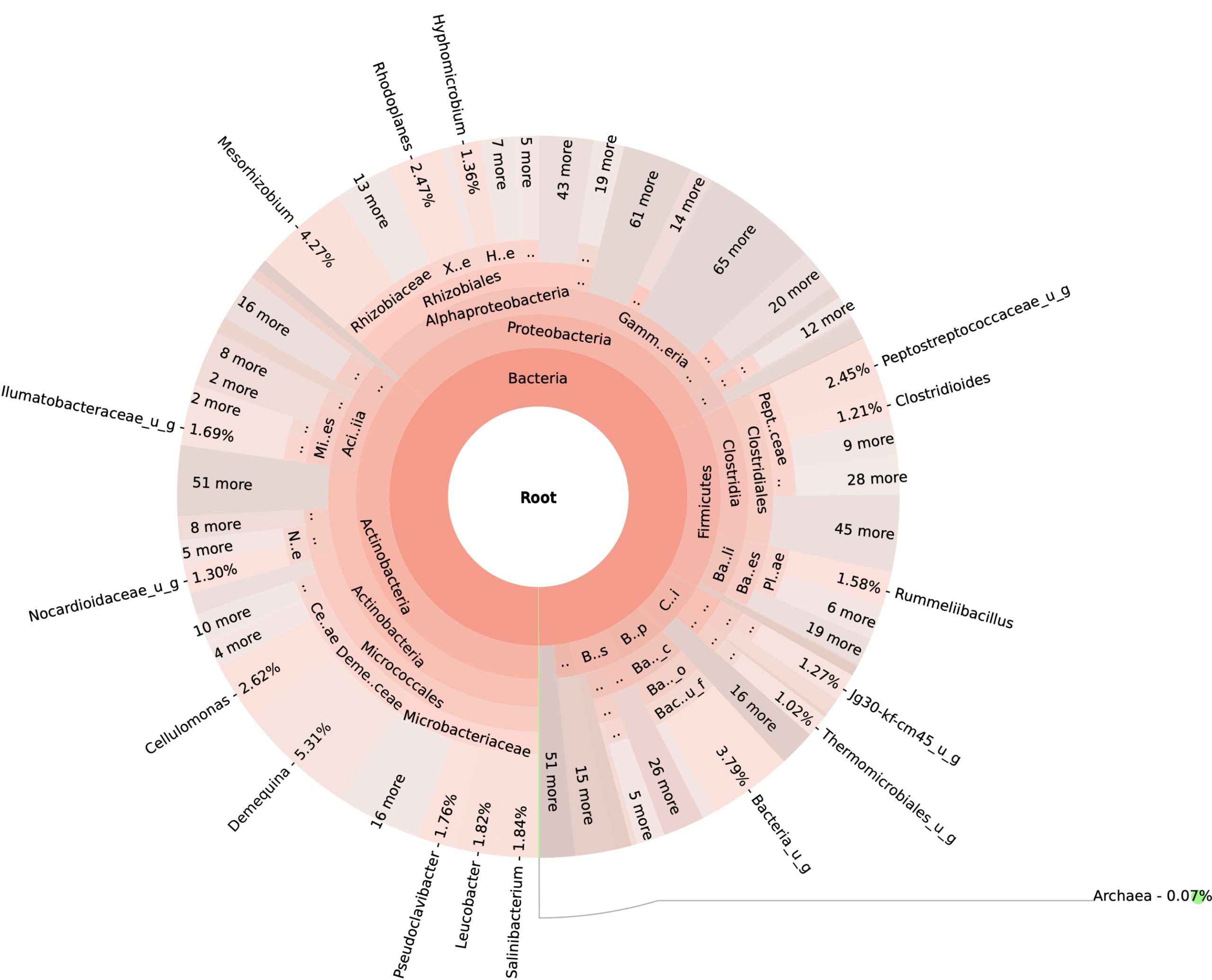

Fig. 3. Sunburst chart analysis depicted the genus level relative abundance across the earthworms’ species: (A) Polypheretima elongata; (B) Perionyx excavatus; (C) Eudrilus eugeniae.
Alpha diversity analysis of earthworms’ gut microbiota
A total 1298 OTUs were obtained during the evaluation of bacterial diversity on CosmosID bioinformatics pipeline (https://app.cosmosid.com). A good coverage sequencing depth (99.8%) was found which represents capturing of majority of the bacterial diversity in all samples. The species richness (Chao1) was highest in Perionyx excavatus (947) followed by Eudrilus eugeniae (751) and lowest in Polypheretima elongata (678). Similar trends were found in Shannon index and Simpson index, the Shannon index for Perionyx excavatus (7.6139) was highest followed by Eudrilus eugeniae (7.2667) and lowest in Polypheretima elongata (5.1776). The Simpson index value for Perionyx excavatus (0.98757) was highest followed by Eudrilus eugeniae (0.98394) and lowest in Polypheretima elongata (0.89856) (Table 2 & Fig. 4A-C). The alpha diversity indices analysis reflected high diversity in Perionyx excavatus followed by Eudrilus eugeniae and lowest in Polypheretima elongata (Fig. 4A-C).
Table (2):
Summary of the bacterial richness and diversity in different earthworm species.
Parameters |
P. excavatus |
E. eugeniae |
P. elongata |
|---|---|---|---|
Total reads |
91938 |
85414 |
77455 |
Good’s coverage % |
99.8 |
99.8 |
99.8 |
Chao1 |
947 |
751 |
678 |
Simpson |
0.98757 |
0.98394 |
0.89856 |
Shannon |
7.6139 |
7.2667 |
5.1776 |
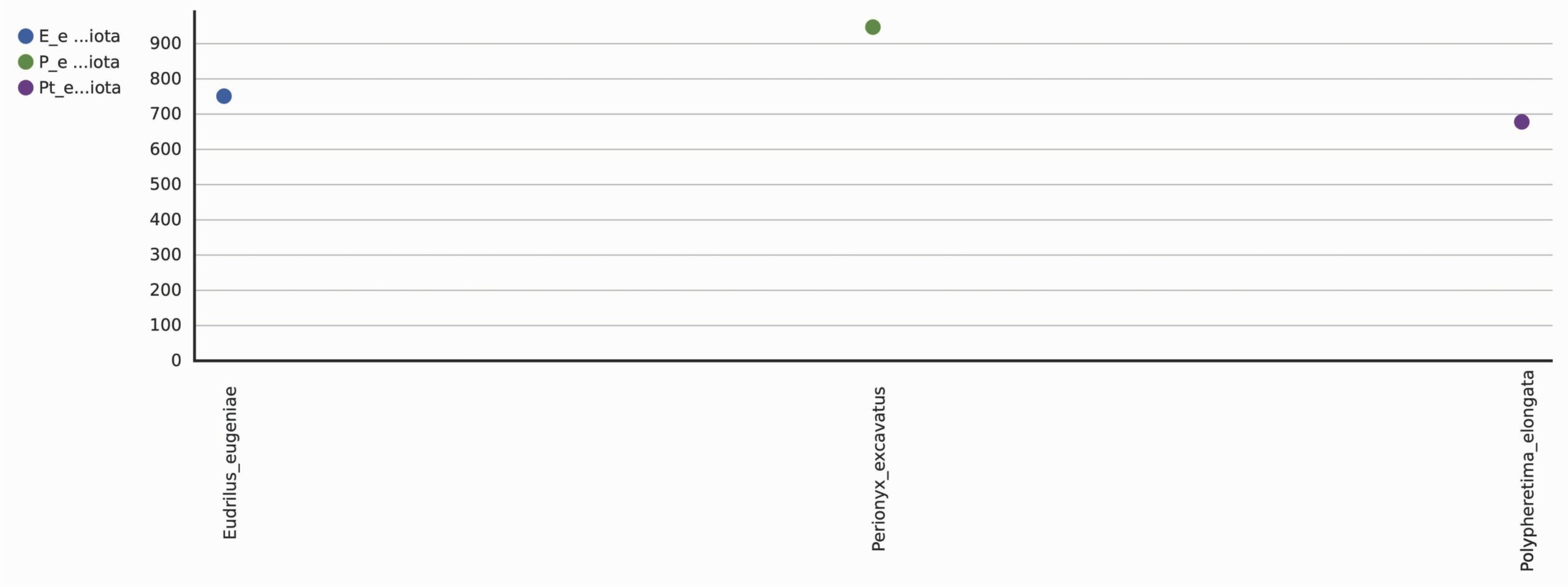
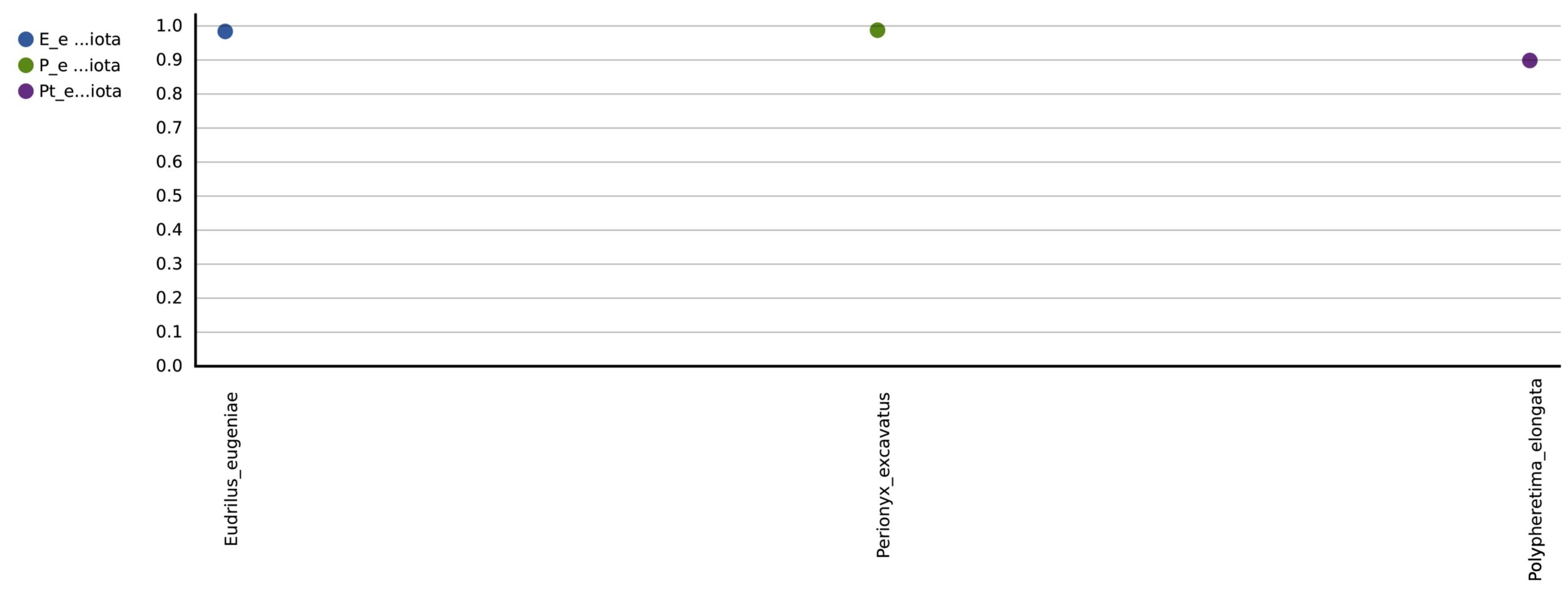
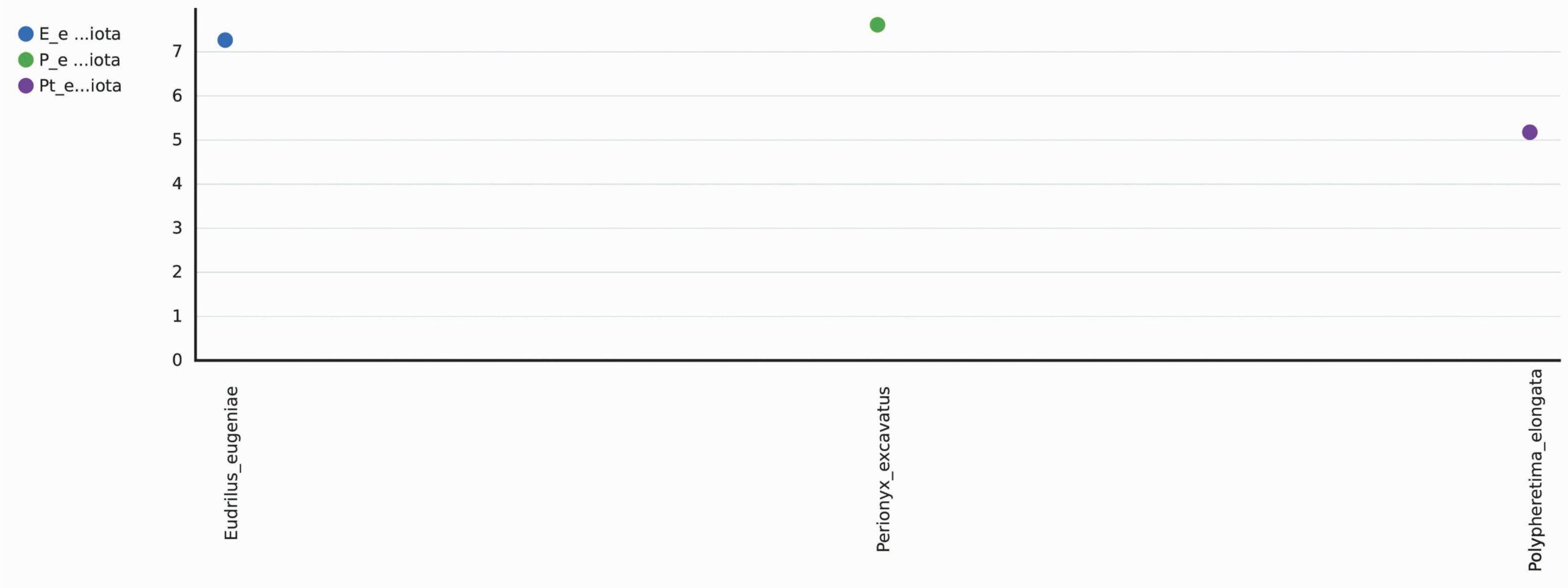
Fig. 4. Alpha diversity analysis of three different earthworms species gut microbiota. (A) Chao1; (B) Simpson index; (C) Shannon index.
Heatmap clustering analysis
The abundance of clusters and similarities were observed by plotting the heatmap, a graphical display of values in colour gradients of data matrix. Where, vertical clustering represents similarity in abundance of different species in the samples. The lesser distances between two samples represents shorter branch lengths that expressed relatively similar abundance between samples. The colour gradient from blue to red symbolizes low to high relative abundance31.
At phylum level
The relative abundance of microbiota in Perionyx excavatus was recorded the highest, followed by Eudrilus eugeniae and the lowest in Polypheretima elongate (Fig. 5A). The upper clustering tree indicates that the bacterial community in Perionyx excavatus and Eudrilus eugeniae, were more similar compare to the Polypheretima elongata.
At genus level
The heatmap analysis of top fifty genera is shown in Fig. 5B. For the top 50 genera, the abundant bacteria in Polypheretima elongata, Perionyx excavatus and Eudrilus eugeniae were barely overlapped (Fig. 5B). Those abundant bacteria in the Polypheretima elongata sample, such as Aeromonas, Enterobacter, Citrobacter, Pseudomonas, Agromyces and Kacuria were relatively low in Perionyx excavatus and Eudrilus eugeniae gut. Some genera including, Mezorhizobium, Rhodoplanes, cellulomonas, Salinibacterium, Pseudoclavibacter, Iamia and leucobacter were abundant in Perionyx excavatus and Eudrilus eugeniae gut. Moreover, Flavobacterium, Arenimonas and Rhodobacter were abundant only in the Eudrilus eugeniae gut. Bradyrhizobium, Hypomicrobium and Rummeliibacillus were abundant only in Perionyx excavatus.
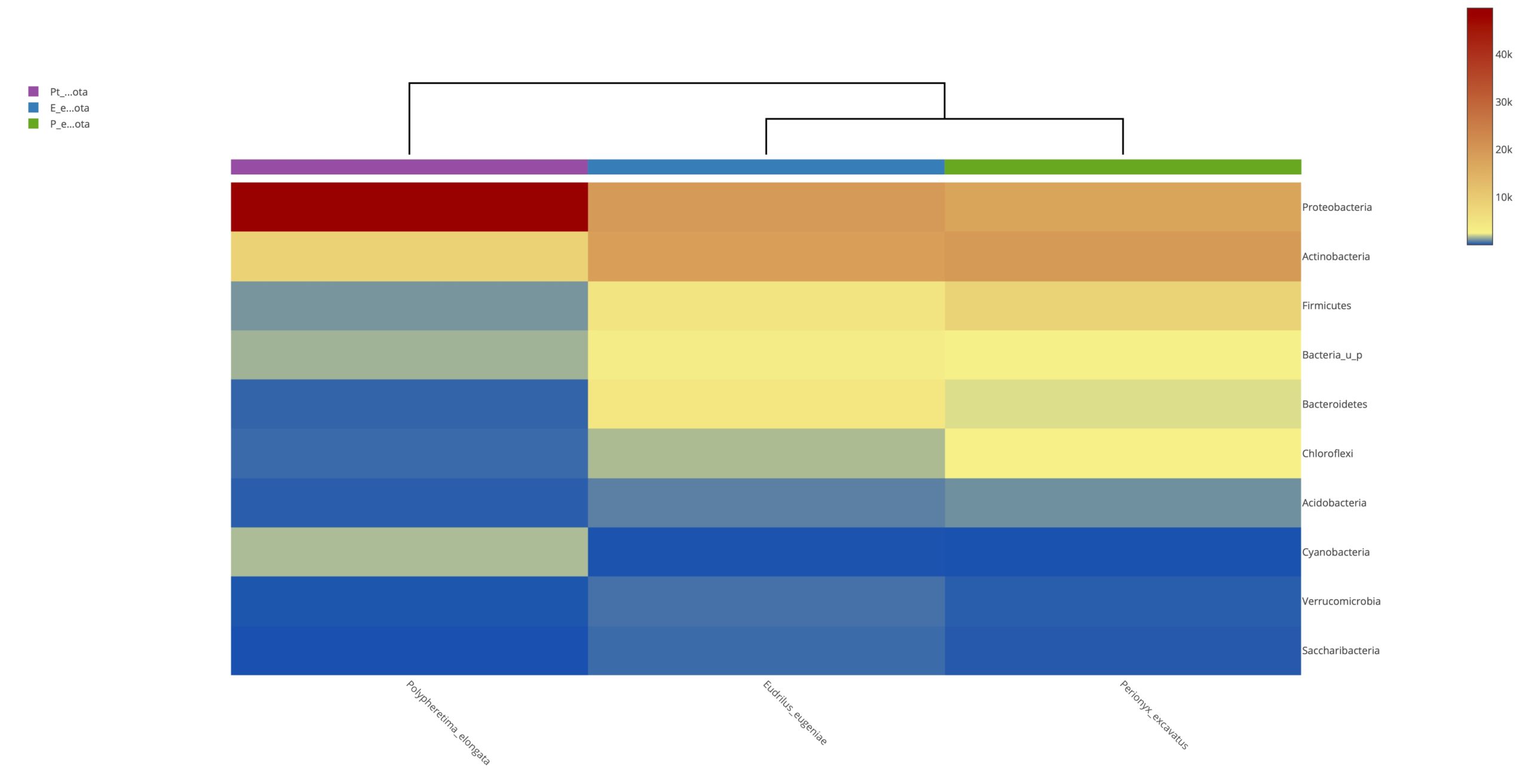
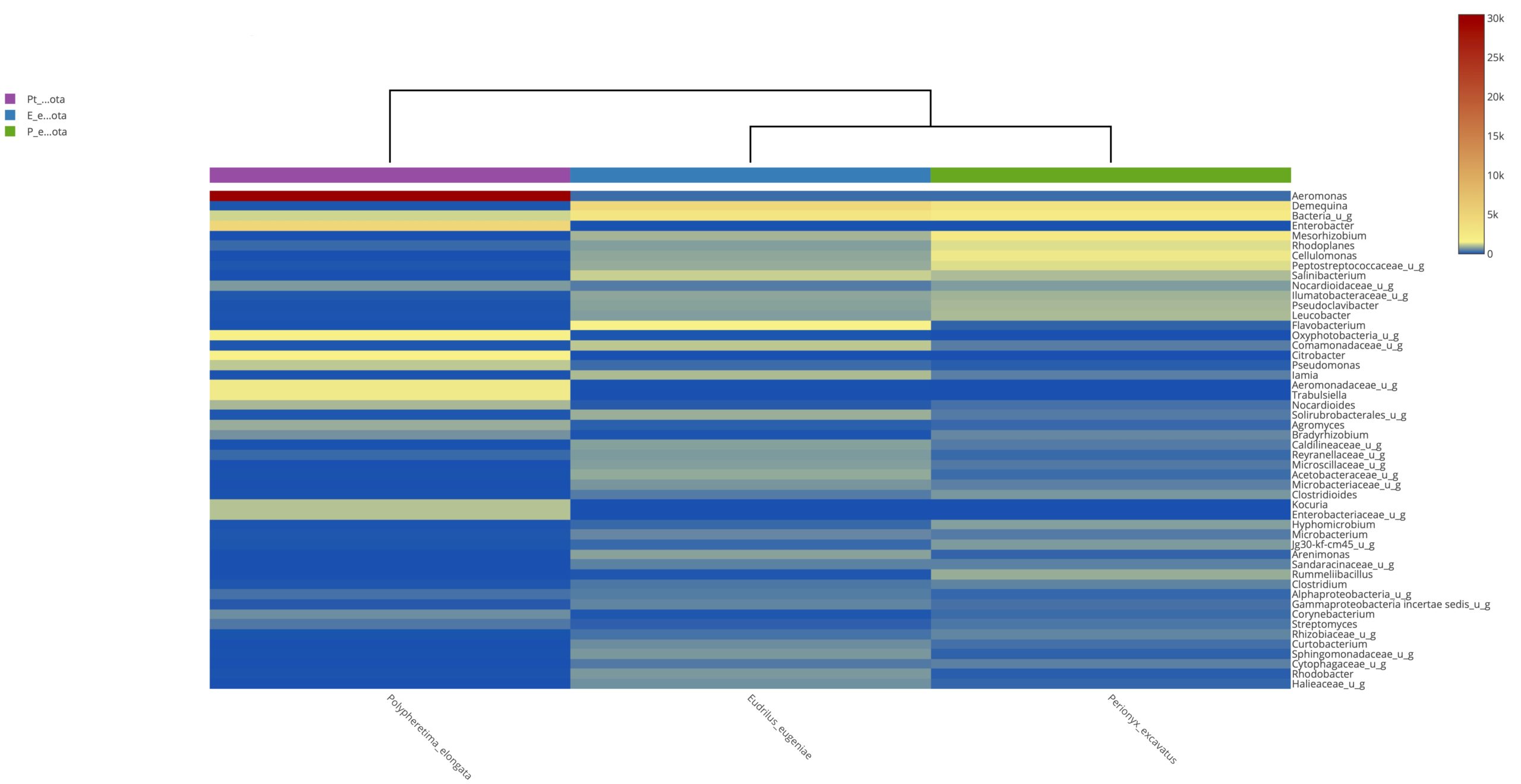
Fig. 5. Heat map of three different earthworms species gut microbiota. (A) at phylum level; (B) at genus level.
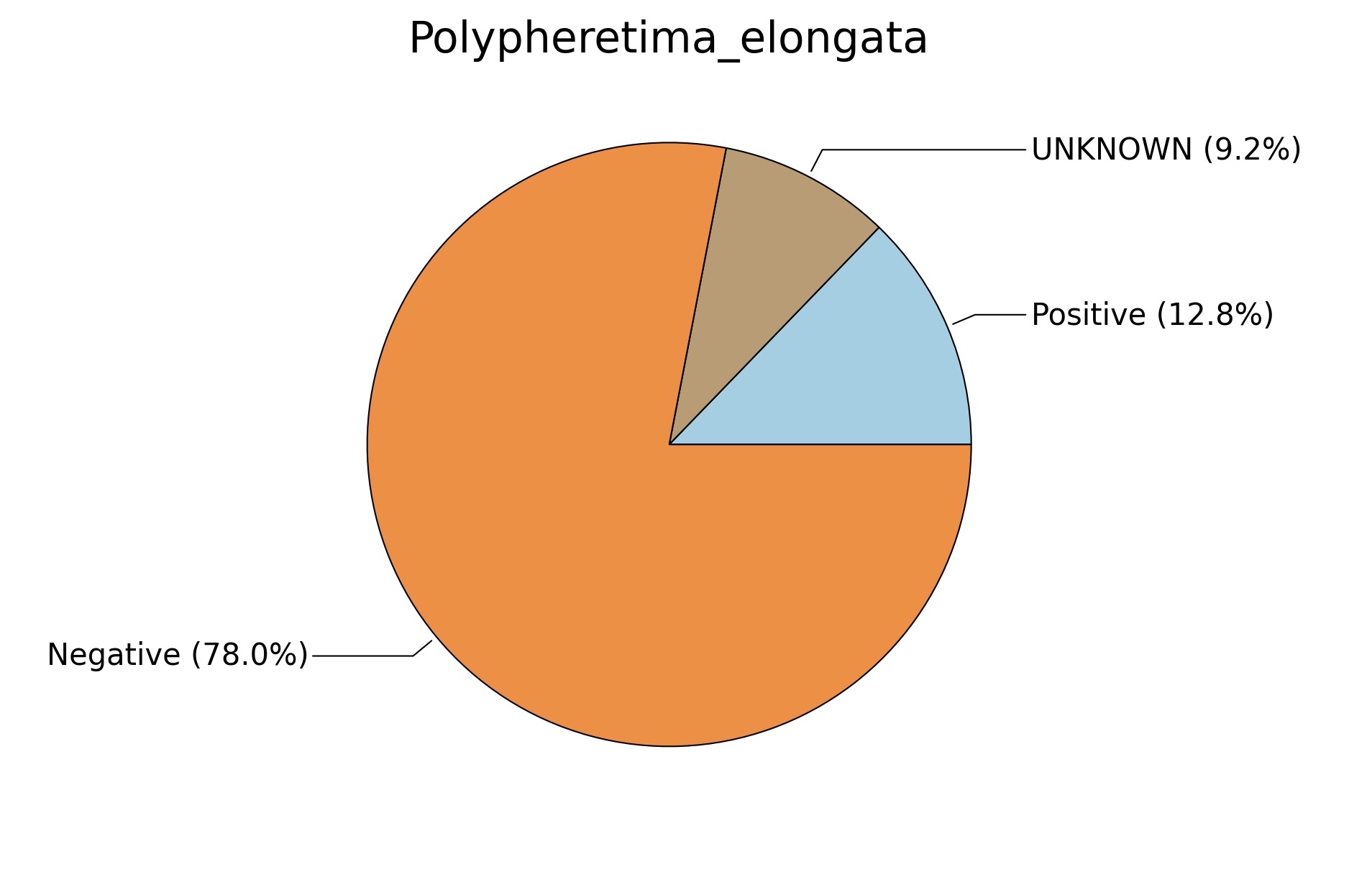

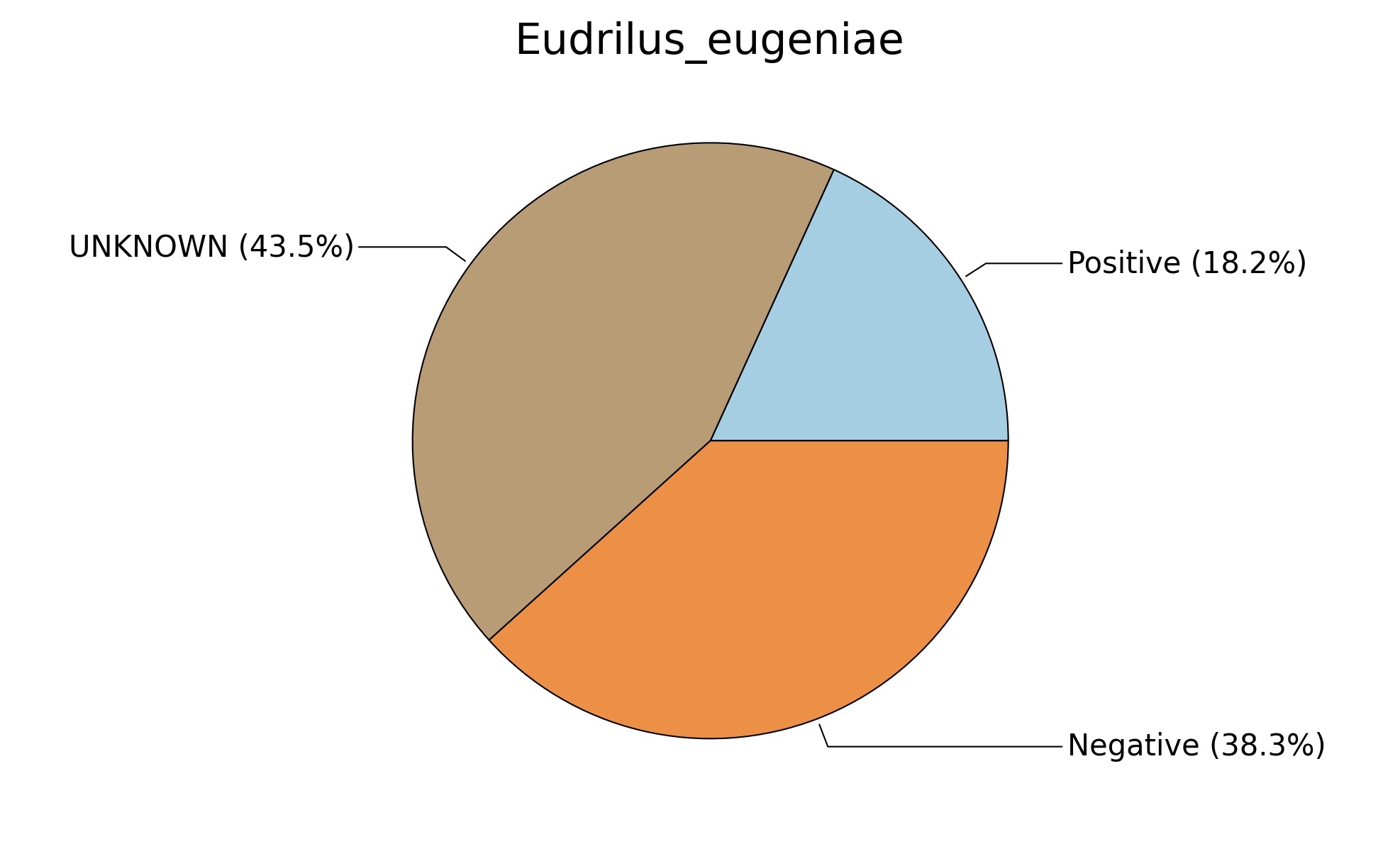
Fig. 6. Phenotypic analysis of three different earthworm sp. gut metagenomes on the basis of Gram staining using METAGENassist, (A) Polypheretima elongata; (B) Perionyx excavatus; (C) Eudrilus eugeniae.
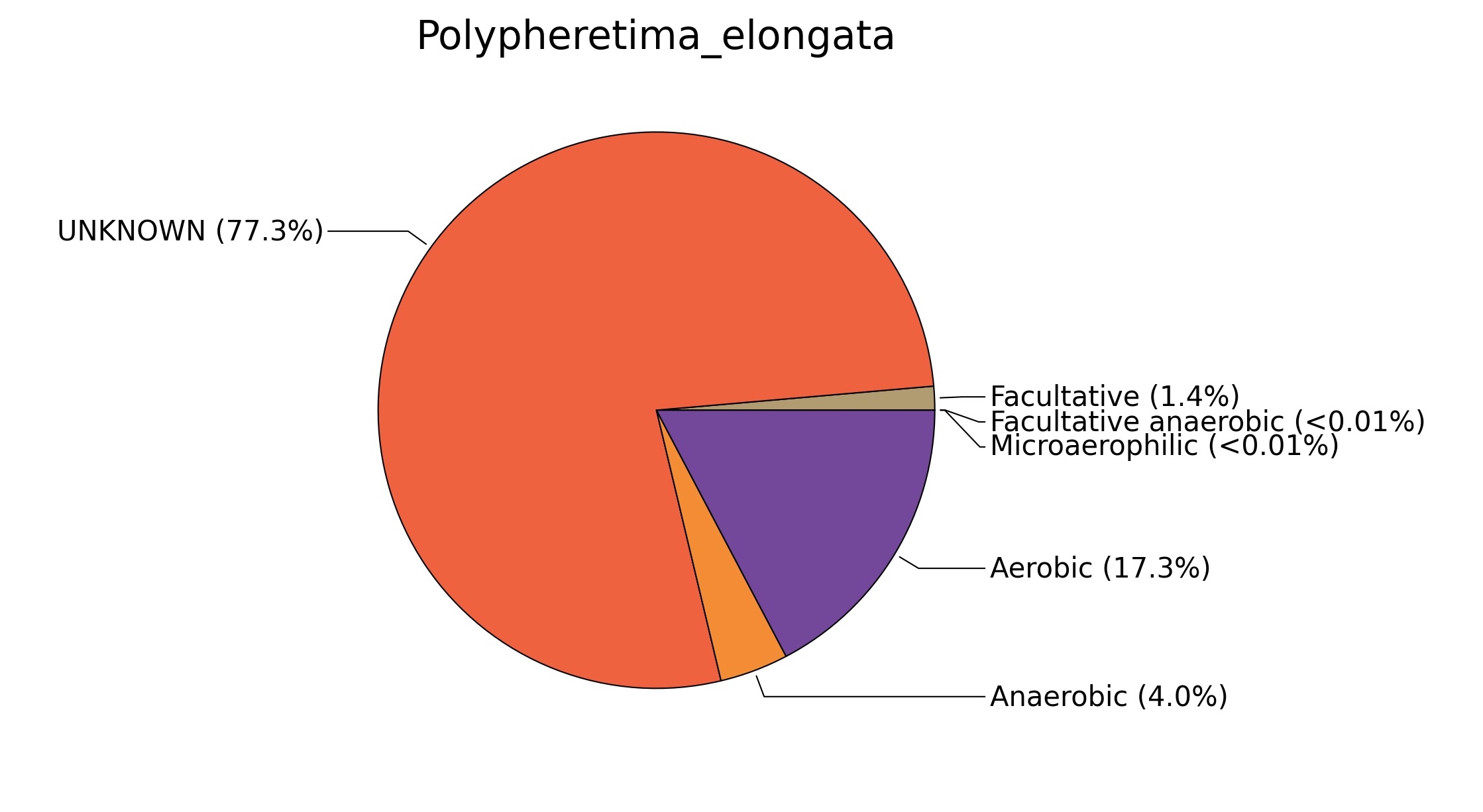
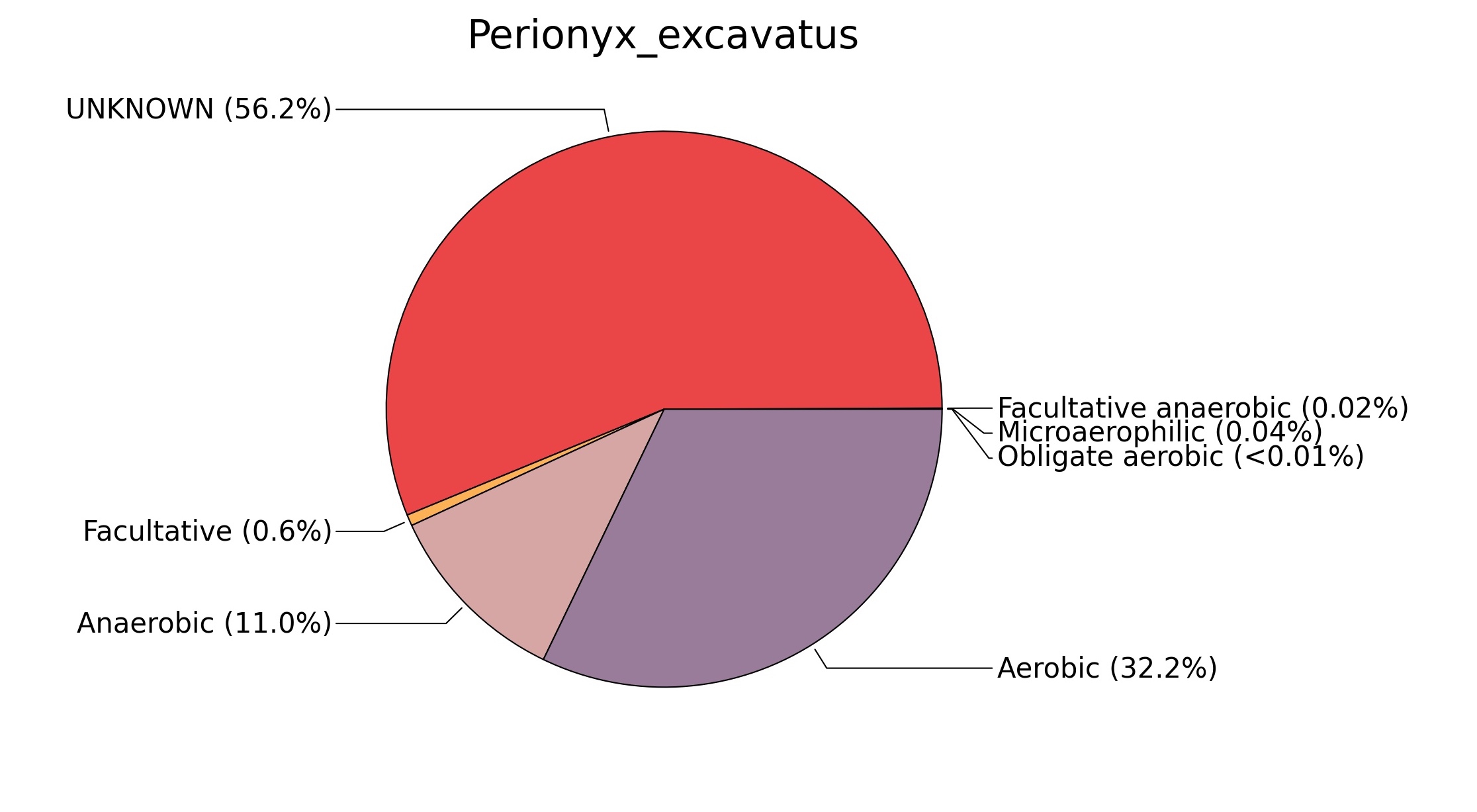
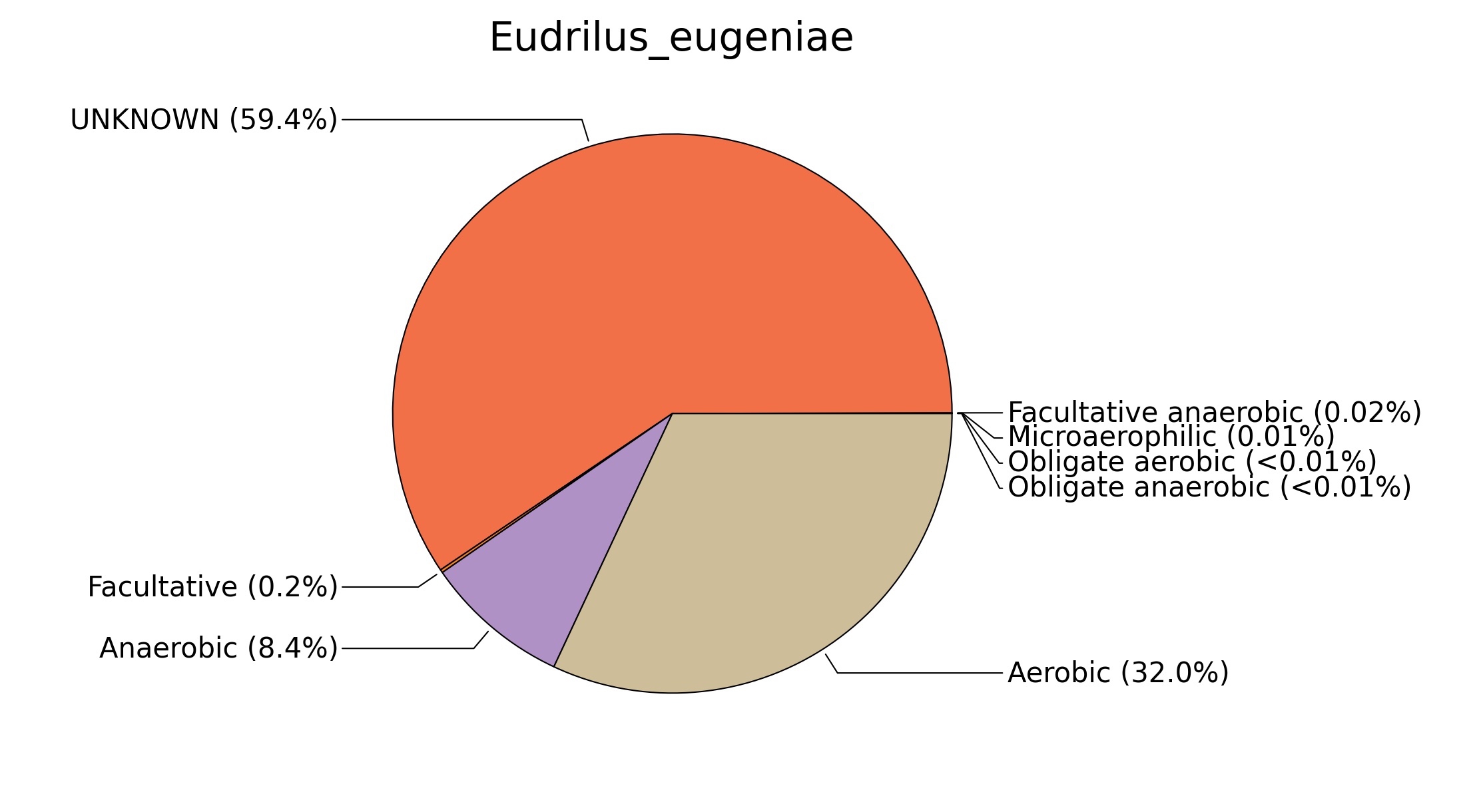
Fig. 7. Phenotypic analysis of three different earthworm sp. gut metagenomes on the basis of oxygen requirement using METAGENassist, (A) Polypheretima elongata; (B) Perionyx excavatus; (C) Eudrilus eugeniae.
Taxonomic to phenotypic profiling
A web-based server METAGENassist was used to map taxonomic to the phenotypic profiling29. Differences were observed in the phenotype classes viz., Gram staining, metabolism and oxygen requirement. On considering Gram staining phenotypic analysis a significant increase in Gram-negative bacteria in Polypheretima elongata gut, was found as compare to Perionyx excavatus and Eudrilus eugeniae gut (Fig. 6A-C). While in case of another phenotypic category oxygen requirement, aerobic followed by anaerobic group were most dominant in all earthworms gut microbiota. It was worthy to note a very high range of unknown sequence reads were found 77.3%, 56.2%, 59.4.4% in Polypheretima elongata, Perionyx excavatus and Eudrilus eugeniae gut microbiota respectively (Fig. 7A-C). The metabolism phenotypic In Silico analysis demonstrated that the gut of earthworm can be looked at a microecological niche, where a number of different biogeochemical cycles, xenobiotic degradation, and lignocellulosic deconstructions are being performed. The taxonomic to the phenotypic characterization of the earthworm gut microbiome from three species showed that bulk of the bacterial communities were ammonia oxidizers in Polypheretima elongata (80.2%), whereas in Perionyx excavatus the relative abundance of ammonia oxidizers was 31.6% and 29.3% in Eudrilus eugeniae. The metagenome of Polypheretima elongata has a relative abundance of sulfate reducer (78.7%), nitrite reducers (73.2%) and chitin degrader (54.1.7%) (Fig. 8A), whereas Perionyx excavatus has a relative abundance of sulfate reducer (25.2%), dehalogenators (25.1%), nitrite reducers (24.2%) and sulphide oxidizer (16.3%) (Fig. 8B). Similarly, Eudrilus eugeniae showed a relative abundance of sulfate reducer (22.6%), dehalogenators (22.0%), nitrite reducers (19.8%) and sulphide oxidizer (13.2%). In the metagenome of Polypheretima elongata, Perionyx excavatus and Eudrilus eugeniae, the relative abundance of nitrogen fixers were 3.4%, 11.2% and 10.2% respectively (Fig. 8A-C).
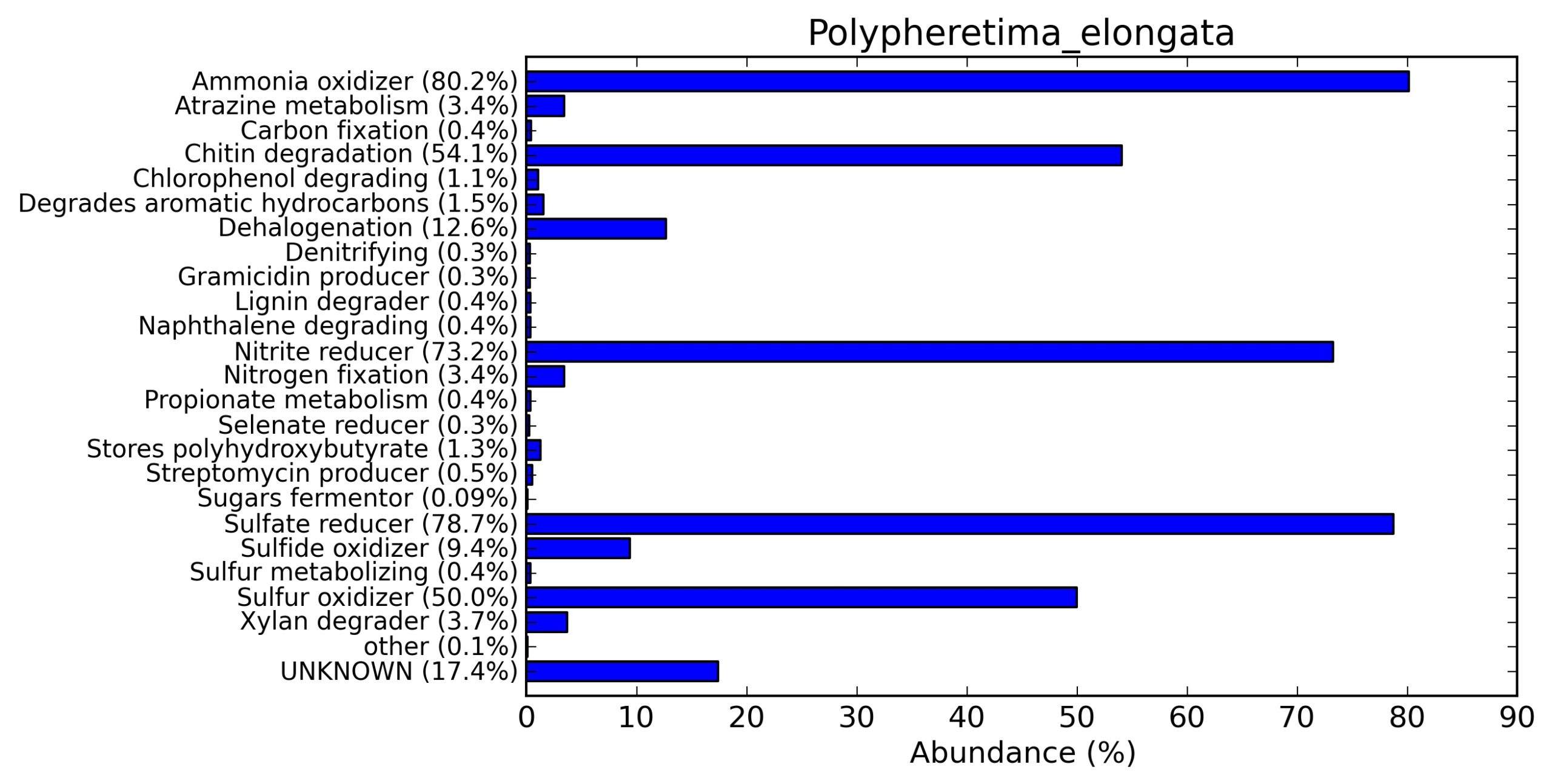

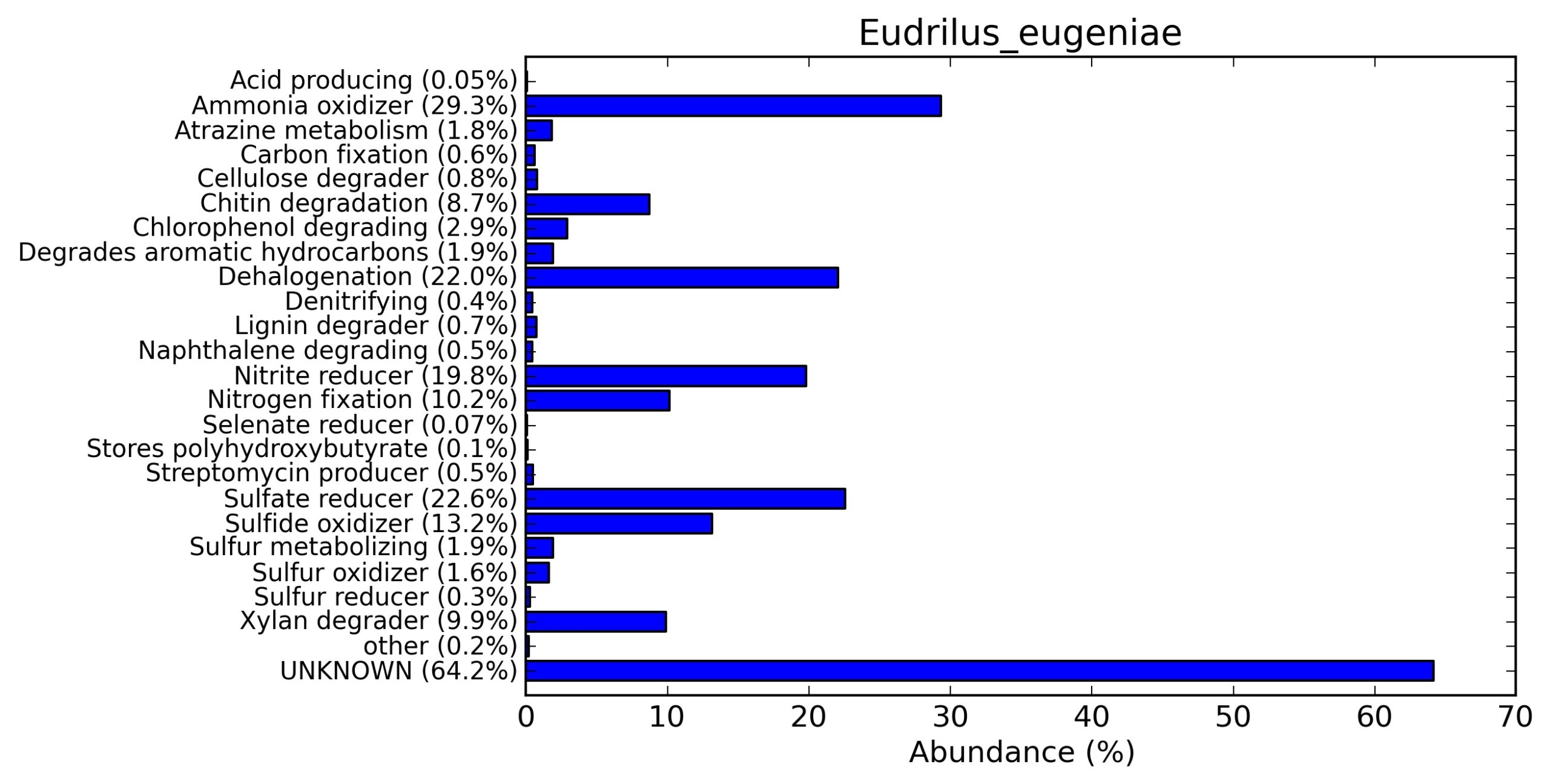
Fig. 8. Phenotypic analysis of three different earthworm sp. gut metagenomes on the basis of functional metabolism using METAGENassist, (A) Polypheretima elongata; (B) Perionyx excavatus; (C) Eudrilus eugeniae.
Earthworms play a vital role in the overall health and maintenance of soil ecosystems by altering soil texture, regulate water content, maintain the availability of nutrients for the plant; this diverse functionality of earthworms is mainly attributed to their gut microbiome32. Recently, scientists are gaining interest on functionality of earthworm gut7,11,33-39 while, the studies on gut associated microbiome are still fragmentary. The present investigation revealed the community structures of gut bacteria of Polypheretima elongata by amplicon sequencing of 16S rRNA gene for the first time, although limited number of studies are available on other earthworm species especially, Eudrilus eugeniae39-42 and Perionyx excavatus with metagenomic pyrosequencing43,44. We recorded a slight divergence in the diversity across the species, e.g the collective reads derived from the Perionyx sp. has the greatest value of Chao1 (947), followed by Eudrilus sp. (751), and Polypheretima sp. (678). Since there was no significant difference found in Good’s coverage value among the samples (> 99%), reflects that sufficient amount of the bacterial diversity were captured in all the samples45-47. The Shannon index ranged from 5.177 to 7.613 and Simpson index ranged from 0.8985- 0.9875 across the samples, indicates Polypheretima elongata had the lowest bacterial diversity, while Perionyx excavatus has the highest diversity, with high species richness. The alpha diversity analysis (which is comprehensive indicator of species richness in community ecology) showed Perionyx excavatus gut had the highest diversity followed by Eudrilus eugeniae while Polypheretima elongata gut was the lowest according to Chao1, Shannon and Simpson values (Table 2 & Fig. 4A-C). The next generation sequences of soil microbiome with respect to soil depth suggested that the upper layer of topsoil have higher microbial diversity than the lower layer of topsoil, with increase in soil depth, microbiome abundance decreases48,49. The topsoil is made up of decomposed material of plants and leaves, which provides a favourable conditions for growth of soil microbes50 and play a crucial role in formation of humus, nutrients and organic matter51. Since epigeic earthworms live and feed in upper layer of topsoil, get high exposure of soil microbiome, humus, nutrient and organic matter. Effect of surrounding environment (available substrate and feeding habit) may not be ignored on counting earthworms’ gut microbiome12,52. Our observation on alpha diversity in gut microbiome of Perionyx excavates, Eudrilus eugeniae and Polypheretima elongata corresponds to above facts. The worms live in the lower layer of topsoil carry relatively low microbial abundance and less availability of nutrients. In addition, alteration in the relative abundance of bacterial communities in earthworms’ gut may be interlinked with variation in their feeding behaviour pattern because of dependence on microbial colonization of host’s feeding behaviour pattern52. The epigeic species Perionyx excavatus and, Eudrilus eugeniae feeding behaviour ranges at upper layer of topsoil, making them access to feed on soil minerals, humus as well as remains of plant materials, while endogeic species Polypheretima elongata feeds on decomposing litter53. The study reported 5 major phyla of bacteria (Proteobacteria, Actinobacteria, Firmicutes, Bacteroidetes and Chloroflexi) in all three earthworm species, irrespective of their different feedings behaviours, which corresponded to the “core microbiota” of earthworms gut, although relative difference were observed at various taxonomical levels when compared for gut microbiota respectively. This could be explained due to co-evolution of certain core taxa via secretions of gut fluids that regulate microbial communities in earthworm gut54, that remains largely unchanged with earthworms12,55. The progression of a particular microbial community depends on the food source and life forms. Moreover, various microbial communities are selected or favoured over other microbes in the tube-like gut, which is stable in the moisture and nutrient conditions, although it acts as unique anoxic micro-environment filtering agent for ingested microbial communities of microorganism pools56.
The gut of Polypheretima elongata, Perionyx excavatus and Eudrilus eugeniae may be viewed as a bioreactor, in which diverse functions (biodegradation, bioremediation and biogeochemical cycling) goes simultaneously. On analysing homology datasets from the various online databases, Proteobacteria was found the predominant phylum, followed by Actinobacteria, Firmicutes and Bacteroidetes. The dominance of Proteobacteria may be due their fast-growing nature and its ability to employ available organic carbon sources and amino acids in the earthworm gut32. The predicted phenotypic analysis showed that gram negative bacteria’s abundance depicts strong relationship between proteobacteria and ammonia oxidizers, sulfate reducers, nitrite reducers and chitin degraders. The nutrient poor environment carries high abundance of protobacteria12,57 which play a vital role in the nitrogen cycle58,59 and cellulose degradation60. It is noteworthy that similar trends have been recorded with predicted phenotype metabolism analysis in earthworms42 and mammals gut microbiome61,62. In addition, few low abundance of certain phyla were also present such as Chloroflexi, Acidobacteria, Saccharibacteria and Verrucomicrobia. The earthworm’s digestive system is involved in various processes such as oxidation and reduction, emission of N2O and N2, remediation, nitrogen fixation, denitrification and degradation processes. Deciphering the earthworm gut microbiome may enable researchers in understanding a much better perspective of their metabolic capabilities. Taxonomic to the phenotypic mapping on the basis of metabolism of the three species suggested that the earthworm gut microbiome played vital role in remediation, denitrification, nitrogen fixation, degradation of cellulose, reduction and sulfur oxidation processes. Isolation of such functionally active bacterial communities from earthworm gut may prove as a pearl of essential enzymes to degrade xenobiotic, lignocellulose for production of biofuels, environmental remediation and biogeochemical cycling.
The present study revealed Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes were the dominant phyla in earthworm sp. Functional characterization revealed that the majority of the bacterial groups were ammonia oxidizers followed by sulfate reducer and nitrite reducer. The study highlights that next-generation high throughput sequencing provides a much detailed and accurate insight into the gut microbiome than other conventional techniques. It can be hypothesized that the majority of the functional attributions of earthworms in the soil ecosystem may be related to their diverse gut microbiome instead the activity of soil microbiomes.
ACKNOWLEDGMENTS
Author SY is thankful to Department of Biotechnology, Ministry of Science and Technology, Govt. of India, New Delhi and SST for Environment Planning and Coordination Organisation (EPCO), Ministry of Environment, Govt. of Madhya Pradesh for the financial support to carry out the study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
SST performed the experiment, recorded and analyzed data with help of available bioinformatics tools and, wrote the manuscript with the support of SKJ and SY. ARL helped in performing experiment and computation of phylogenomic. NT helped in the draft preparation. All authors provided critical feedback and helped to shape the research, analysis, and manuscript.
FUNDING
Author (SY) received a financial grant under the project entitled Toxicity of NPs against earthworm and investigating its potential as bio-transforming agent with Grant ID: BT/PR1856/NNT/28/532/2011 from the Department of Biotechnology, Ministry of Science and Technology, Govt. of India.
ETHICS STATEMENT
This article does not contain any studies with human participants or animals performed by any of the authors.
AVAILABILITY OF DATA
The 16S rRNA amplicon sequences of earthworms gut were submitted in NCBI sequences read archive under the bio project SRA accession number PRJNA670329.
- Pass DA, Morgan AJ, Read DS, Field D, Weightman AJ, Kille P. The effect of anthropogenic arsenic contamination on the earthworm microbiome. Environ Microbiol. 2015;17(6):1884-1896.
Crossref - Elyamine A, Afzal J, Rana M, Imran M, Cai M, Hu C. Phenanthrene Mitigates Cadmium Toxicity in Earthworms Eisenia fetida (Epigeic Species) and Aporrectodea caliginosa (Endogeic Species) in Soil. Int J Environ Res Public Health. 2018;15(11):2384.
Crossref - Bardgett RD, Chan KF. Experimental evidence that soil fauna enhance nutrient mineralization and plant nutrient uptake in montane grassland ecosystems. Soil Biol Biochem. 1999;31(7):1007-1014.
Crossref - Blouin M, Hodson ME, Delgado EA, et al. A review of earthworm impact on soil function and ecosystem services. European J Soil Sci. 2013;64(2):161-182.
Crossref - Lubbers IM, Van Groenigen KJ, Fonte SJ, Six J, Brussaard L, Van Groenigen JW. Greenhouse-gas emissions from soils increased by earthworms. Nat Climate Change. 2013;3(3):187-194.
Crossref - Andriuzzi WS, Ngo PT, Geisen S, et al. Organic matter composition and the protist and nematode communities around anecic earthworm burrows. Biol Fertility of Soils. 2016;52(1):91-100.
Crossref - Thakuria D, Schmidt O, Finan D, Egan D, Doohan FM. Gut wall bacteria of earthworms: a natural selection process. ISMEJ. 2010;4(3):357-366.
Crossref - Van Groenigen JW, Lubbers IM, Vos HM, Brown GG, De Deyn GB, Van Groenigen K. Earthworms increase plant production: a meta-analysis. Sci. Rep. 2014;4:6365.
Crossref - Le Bayon RC, Bullinger-Weber G, Schomburg A, Turberg P, Schlaepfer R, Guenat C. Earthworms as ecosystem engineers: a review. Earthworms: Types, Roles and Research. In: Horton CG, Ed. NOVA Science Publishers, New York, 2017:129-177.
- Dishaw LJ, Cannon JP, Litman GW, Parker WJD. Immune-directed support of rich microbial communities in the gut has ancient roots. Dev Comp Immunol. 2014;47(1):36-51.
Crossref - Ma L, Xie Y, Han Z, Giesy JP, Zhang X. Responses of earthworms and microbial communities in their guts to Triclosan. Chemosphere. 2017;168:1194-1202.
Crossref - Liu D, Lian B, Wu C, Guo P. A comparative study of gut microbiota profiles of earthworms fed in three different substrates. Symbiosis. 2018;74(1):21-29.
Crossref - Bouché MB. Stratégies lombriciennes. In Lohm U, Persson E (eds.), Soil Organisms as Components of Ecosystems. Ecological Bulletins, Swedish Natural Science Research Council, Stockholm, 1977:122–132.
- Knapp B, Podmirseg S, Seeber J, Meyer E, Insam H. Diet-related composition of the gut microbiota of Lumbricus rubellus as revealed by a molecular fingerprinting technique and cloning. Soil Biol Biochem. 2009;41(11):2299-2307.
Crossref - Julka JM, Paliwal R. Seasonal changes in the population of earthworms (Oligochaeta) in an orchard. J Bombay Nat Hist Soc. 1990;87:323-326.
- Thakur SS, Lone AR, Tiwari N, Jain SK, James SW, Yadav S. A contribution to the earthworm diversity (Clitellata, Moniligastridae) of Kerala, a component of the Western Ghats biodiversity hotspot, India, using integrated taxonomy. Animal Biodiversity and Conservation. 2021;44(1):117-137.
Crossref - Kimura M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J Mol Evol. 1980;16(2):111-120.
Crossref - Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32(5):1792-1797.
Crossref - Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol. 2018;35(6):1547-1549.
Crossref - Klindworth A, Pruesse E, Schweer T, et al. Evaluation of general 16S ribosomal RNA gene PCR primers for classical and next-generation sequencing-based diversity studies. Nucleic Acids Res. 2013;41(1):e1.
Crossref - Caporaso JG, Lauber CL, Walters WA, et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISMEJ. 2012;6(8):1621-1624.
Crossref - Andrews S. FastQC: a quality control tool for high throughput sequence data 2010. http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ Accessed 20 November 2020.
- Lax S, Smith DP, Hampton-Marcell J, et al. Longitudinal analysis of microbial interaction between humans and the indoor environment. Science. 2014;345(6200):1048-1052.
Crossref - Ponnusamy D, Kozlova EV, Sha J, et al. Cross-talk among flesh-eating Aeromonas hydrophila strains in mixed infection leading to necrotizing fasciitis. Proc Natl Acad Sci. 2016;113(3):722-727.
Crossref - Janes VA, Matamoros S, Willemse N, et al. Metagenomic sequencing to replace semi-quantitative urine culture for detection of urinary tract infections: a proof of concept. bioRxiv. 2017. Accessed 28 November 2020.
Crossref - Junqueira ACM, Ratan A, Acerbi E, et al. The microbiomes of blowflies and houseflies as bacterial transmission reservoirs. Sci Rep. 2017;7:16324.
Crossref - Connelly S, Bristol JA, Hubert S, et al. SYN-004 (ribaxamase), an oral beta-lactamase, mitigates antibiotic-mediated dysbiosis in a porcine gut microbiome model. J appl Microbiol. 2017;123(1):66-79.
Crossref - Yan Q, Wi YM, Thoendel MJ, et al. Evaluation of the CosmosID Bioinformatics Platform for Prosthetic Joint-Associated Sonicate Fluid Shotgun Metagenomic Data Analysis. J Clin Microbiol. 2019;57(2):e01182-18.
Crossref - Arndt D, Xia J, Liu Y, et al. METAGENassist: a comprehensive web server for comparative metagenomics. Nucleic Acids Res. 2012;40(1):88-95.
Crossref - Paul SS, Chatterjee RN, Raju MVLN, et al. Gut Microbial Composition Differs Extensively among Indian Native Chicken Breeds Originated in Different Geographical Locations and a Commercial Broiler Line, but Breed-Specific, as Well as Across-Breed Core Microbiomes, Are Found. Microorganisms. 2021;9(2):391.
Crossref - Wilkinson L, Friendly M. The history of the cluster heat map. The American Statistician. 2009;63(2):179-184.
Crossref - Medina-Sauza RM, Alvarez-Jimenez M, Delhal A, et al. Earthworms building up soil microbiota, a review. Front Environ Sci. 2019;7(81):1-20.
Crossref - Rattray RM, Perumbakkam S, Smith F, Craig AM. Microbiomic comparison of the intestine of the earthworm Eisenia fetida fed ergovaline. Curr Microbiol. 2010;60(3):229-235.
Crossref - Depkat-Jakob PS, Brown GG, Tsai SM, Horn MA, Drake HL. Emission of nitrous oxide and dinitrogen by diverse earthworm families from Brazil and resolution of associated denitrifying and nitrate-dissimilating taxa. FEMS Microbiol Ecol. 2013;83(2):375-391.
Crossref - Dvorak J, Mancikova V, Pizl V, et al. Microbial environment affects innate immunity in two closely related earthworm species Eisenia andrei and Eisenia fetida. PloS One. 2013;8(11):e79257.
Crossref - Aira M, Bybee S, Perez-Losada M, Dominguez J. Feeding on microbiomes: effects of detritivore on the taxonomic and phylogenetic bacterial composition of animal manures. FEMS Microbiol Ecol. 2015;91(11):117.
Crossref - Ding J, Zhu D, Hong B, et al. Long-term application of organic fertilization causes the accumulation of antibiotic resistome in earthworm gut microbiota. Environ Int. 2019;124:145-152.
Crossref - Shi Z, Wang C, Zhao Y. Effects of surfactants on the fractionation, vermiaccumulation, and removal of fluoranthene by earthworms in soil. Chemosphere. 2020;250:126332.
Crossref - Krishnaswamy VG, Jaffar MF, Sridharan R, et al. Effect of chlorpyrifos on the earthworm Eudrilus euginae and their gut microbiome by toxicological and metagenomic analysis. World J Microbiol Biotechnol. 2021;37:76.
Crossref - Schulz K, Hunger S, Brown GG, et al. Methanogenic food web in the gut contents of methane-emitting earthworm Eudrilus eugeniae from Brazil. ISME J. 2015;9(8):1778-1792.
Crossref - Ekperusi OA, Aigbodion IF. Bioremediation of heavy metals and petroleum hydrocarbons in diesel contaminated soil with the earthworm: Eudrilus eugeniae. Springer Plus. 2015;4:540.
Crossref - Owagboriaye F, Dedeke G, Bamidele J, et al. Biochemical response and vermiremediation assessment of three earthworm species (Alma millsoni, Eudrilus eugeniae and Libyodrilus violaceus) in soil contaminated with a glyphosate-based herbicide. Ecol Indicators. 2020;108:105678.
Crossref - Singh A, Singh DP, Tiwari R, et al. Taxonomic and functional annotation of gut bacterial communities of Eisenia foetida and Perionyx excavatus. Microbiol Res. 2015;175:48-56.
Crossref - Singh A, Tiwari R, Sharma A, et al. Taxonomic and functional diversity of the culturable microbiomes of epigeic earthworms and their prospects in agriculture. J Basic Microbiol. 2016;56(9):1009-1020.
Crossref - Zhang J, Chen L, Xing F, et al. Extensive sequence divergence between the reference genomes of two elite indica rice varieties Zhenshan 97 and Minghui 63. Proc Nat Acad Sci. 2016;113(35):5163-5171.
Crossref - Xu S, Yao J, Ainiwaer M, Hong Y, Zhang Y. Analysis of bacterial community structure of activated sludge from wastewater treatment plants in winter. BioMed Res Int. 2018;2018;8278970.
Crossref - Qingqing B, Jie Z, Songben Q, Juan C, Lei Z, Mu X. Cervicovaginal microbiota dysbiosis correlates with HPV persistent infection. Microb Pathog. 2021;152:104617.
Crossref - Celestina C, Wood JL, Manson JB, et al. Microbial communities in top-and subsoil of repacked soil columns respond differently to amendments but their diversity is negatively correlated with plant productivity. Sci Rep. 2019;9:8890.
Crossref - Hao J, Chai YN, Lopes LD, et al. The Effects of Soil Depth on the Structure of Microbial Communities in Agricultural Soils in Iowa, USA. Appl Environ Microbiol. 2021;87(4):e02673-20.
Crossref - Vorholt JA. Microbial life in the phyllosphere. Nat Rev Microbiol. 2012;10(12):828-840.
Crossref - Jacoby R, Peukert M, Succurro A, Koprivova A, Kopriva S. The role of soil microorganisms in plant mineral nutrition-current knowledge and future directions. Front Plant Sci. 2017;8:1617.
Crossref - Sapkota R, Santos S, Farias P, Krogh PH, Winding A. Insights into the earthworm gut multi-kingdom microbial communities. Sci Total Environ. 2020;727:138301.
Crossref - Schmidt O, Scrimgeour CM, Handley LL. Natural abundance of 15N and 13C in earthworms from a wheat and a wheat-clover field. Soil Biol Biochem. 1997:29(9-10):1301-1308.
Crossref - Byzov BA, Khomyakov NV, Kharin SA, Kurakov AV. Fate of soil bacteria and fungi in the gut of earthworms. European J Soil Biol. 2007;43:S149-S156.
Crossref - Srut M, Menke S, Hockner M, Sommer S. Earthworms and cadmium-Heavy metal resistant gut bacteria as indicators for heavy metal pollution in soils? Ecotoxicol Environ Saf. 2019;171:843-853.
Crossref - Drake HL, Horn MA. As the worm turns: the earthworm gut as a transient habitat for soil microbial biomes. Annu Rev Microbiol. 2007;61:169-189.
Crossref - Regar RK, Gaur VK, Bajaj A, Tambat S, Manickam N. Comparative microbiome analysis of two different long-term pesticide contaminated soils revealed the anthropogenic influence on functional potential of microbial communities. Sci Total Environ. 2019;681:413-423.
Crossref - Amoo AE, Babalola OO. Ammonia-oxidizing microorganisms: key players in the promotion of plant growth. J Soil Sci Plant Nutrition. 2017;17(4):935-947.
Crossref - Baskaran V, Patil PK, Antony ML, et al. Microbial community profiling of ammonia and nitrite oxidizing bacterial enrichments from brackishwater ecosystems for mitigating nitrogen species. Sci Rep. 2020;10:5201.
Crossref - Tian JH, Pourcher AM, Bouchez T, Gelhaye E, Peu P. Occurrence of lignin degradation genotypes and phenotypes among prokaryotes. Appl Microbiol Biotechnol. 2014;98(23):9527-9544.
Crossref - Yasuda K, Oh K, Ren B, et al. Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microbe. 2015;17(3):385-391.
Crossref - Vemuri R, Sherrill C, Davis M, Kavanagh K. Age-Related Colonic Mucosal Microbiome Community Shifts in Monkeys. J Gerontol A Biol Sci Med Sci. 2020;glaa256.
Crossref
© The Author(s) 2021. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.