


ISSN: 0973-7510
E-ISSN: 2581-690X
Symbiosis is a complex genetic regulatory biological evolution which is highly specific pertaining to plant species and microbial strains. Biological nitrogen fixation in legumes is a functional combination of nodulation by nod genes and regulation by nif, fix genes. Three rhizobial strains (Rhizobium leguminosarum, Bradyrhizobium japonicum, and Mesorhizobium ciceri) that we considered for in silico analysis of nif A are proved to be the best isolates with respect to N2 fixing for ground nut, chick pea and soya bean (in vitro) out of 47 forest soil samples. An attempt has been made to understand the structural characteristics and variations of nif genes that may reveal the factors influencing the nitrogen fixation. The primary, secondary and tertiary structure of nif A protein was analyzed by using multiple bioinformatics tools such as chou-Fasman, GOR, ExPasy ProtParam tools, Prosa -web. Literature shows that the homology modeling of nif A protein have not been explored yet which insisted the immediate development for better understanding of nif A structure and its influence on biological nitrogen fixation. In the present predicted 3D structure, the nif A protein was analyzed by three different software tools (Phyre2, Swiss model, Modeller) and validated accordingly which can be considered as an acceptable model. However further in silico studies are suggested to determine the specific factors responsible for nitrogen fixing in the present three rhizobial strains.
Nif A protein; In silico analysis; Nitrogen fixation; Bioinformatic tools; Homology modeling.
Plant microbial interaction is an ever expanding domain in the ecosystem. Circadian emergence and exploration of novel species day by day is broadening the scope and pertinence of microorganism. One such well ascertained biological process that persists through ages in science is symbiosis. Symbiosis perhaps is a complex and differentiating process that unveil its functional specificity pertaining to evolution and exploration of its partnering rhizobial strains1. This could be the probable reason why understanding symbiosis is a perpetual research.
The typical process of nitrogen fixing is facilitated and regulated by three important genes i.e. Nif, Nod and Fix genes with the aid of rhizobium in the nodules of leguminous plants2. Nif genes are diversified and unusually found in nitrogen fixing bacteria. Nitrogen fixation is a complex mechanism; not any single gene involved in the whole process but there are several nif genes with their specific function in nitrogen fixation, assimilation and regulation3. These nif genes are also found on symbiotic bacterial plasmids along with nod genes4. Nif genes code for proteins that are essential to fix and regulate nitrogen in legumes; nitrogenase being one among them5. In most of the diazotroph organisms, the nitrogen fixation genes (Nif) transcription is driven by RNA polymerase which is an alternative holoenzyme and also have a need of nif A activator protein. Environmental effectors usually regulate the activity and synthesis of nif A genes. Oxygen and ammonia are the two major signals which regulate the nitrogen fixation at the extent of nif genes6.
Nif A plays a major role in transcriptional activation and controls the expression of nitrogenase structural genes, genes encoding accessory functions with the association of RNA-polymerase sigma factor Rpo N7. Bacterial conversion of Nitrogen (N2) to ammonia (NH3) an energetically expensive process and very sensitive to oxygen (O2)8. To create a favorable environment within the nodule tissue a specialized plant cells acts as oxygen barriers. Furthermore nodulin, leghemoglobin makes the low oxygen concentration by reversibly binding the oxygen. In bacteria transcription of nitrogen fixing genes largely induced at low oxygen levels9. Under reducing, nitrogen-limiting conditions, NifA is released from NifL to activate transcription at nif promoters.
In vitro analysis of Rhizobium leguminosarum, Bradyrhizobium japonicum, Mesorhizobium Ciceri in ground nut, soya bean and chick pea respectively showed highest plant growth when compared to the rest of the rhizobacteria. Biochemical tests for the respective root nodules showed elevated levels of nitrogen in all the three Rhizobium leguminosarum, Bradyrhizobium japonicum, Mesorhizobium Ciceri10. Further molecular analysis of nif genes by polymerase chain reaction (PCR) showed prominent appearance of nif A band where as other nif genes are with faint or no bands (data not shown). With the evidence of in vitro studies (elevated levels of nitrogen and ACC, IAA plant growth hormones) we further extended the research to understand the role of nif A genes in nitrogen fixation by using in silico model.
The availability of structural model of a protein is one of the keys for understanding biological processes at a molecular level. However, very little is known about the structure and role of nif A proteins. Identification of the 3D structure of a protein is very difficult and complex assignment. Generally two techniques X-ray crystallography or NMR (Nuclear Magnetic Resonance) are used, which are time consuming and expensive11. In this regard, a viable alternative approach is to predict the in silico 3D structure of proteins based on homology modeling technique serves the purpose with better validation. Homology Modeling is known to be one of the best and extensively used methods where in the alignment of know protein structures (templates) was done with the unknown protein sequence which has more than 35% of similarity12.
Sequences of all Nif A proteins are roughly of similar lengths, varying between 519 (R. leguminosarum) and 605 (B. japonicum) amino acids, except that of M. Ciceri, which has only 352 amino acid. Besides, in most rhizobia the nif A gene is subjected to transcriptional regulation although the mechanisms vary depending on the rhizobial strain. Nif A is a three-domain protein13, with a central domain of about 220 amino acids which is sufficient by itself to activate transcription14. The N-terminal domain function is unknown in Nif A, and is absent in M. Ciceri. Whereas the C-terminal domain contains a helix–turn–helix motif that helps in binding to the upstream activator sequence (UAS)15,16. Since the central domain plays a major role in activating the promoter region of nif A, the sequences of the central domains of B. japonicum, R. leguminosarum, M. Ciceri were compared. For better explanation of the mechanism behind nitrogen fixation we checked the in silico protein structural variation and conserved amino acid sequences by modeling primary, secondary and tertiary structures. However, tertiary structures of large number of nitrogenase proteins from different diazotrophs particularly those of symbiotic ones has not yet been resolved. Therefore, there is a need to model a tertiary structure of the Nif A for further understanding of transcriptional activity
Nif-A Protein Sequences of Rhizobia
The nif A protein sequences of B. japonicum, R. leguminosarum, M. Ciceri were retrieved from Uniprot17, a freely accessible resource of protein sequence and functional information (Table 1). The Accession No: for each organism was Q9AMY3 for B. japonicum, P09828 for R. leguminosarum and A0A165VD05 for M. ciceri.
Table (1):
The Protein sequence retrieved from the UniProt
Gene name |
Length modelled in complete sequence |
Uniprot Id |
Organism |
|---|---|---|---|
Nif A |
253-601 |
Q9AMY3 |
Bradyrhizobium japonicum |
Nif A specific regulatory protein |
177-519 |
P09828 |
Rhizobium leguminosarum |
Transcriptional regulator Nif A |
1-351 |
A0A165VD05 |
Mesorhizobium Ciceri |
Physico-Chemical Characteristics
To analyze the physical and chemical characteristics such as molecular weight, theoretical pI, amino acid composition, atomic composition, extinction coefficient, estimated half life, instability index, aliphatic index, and Grand Average of Hydropathicity (GRAVY) of the nif A protein, was computed by ProtParam tool18 (table 2).
Table (2):
Physicochemical properties of Nif A protein.( M. wt.: Molecular weight; pI: Isoelectric point; –R: Number of negative residues; +R: Number of positive residues; EC: Extinction coefficient at 280 nm; II: Instability index; AI: Aliphatic index; GRAVY: Grand Average Hydropathy)
S.No |
Name of the Organism |
M. Wt. |
Seq. Length |
pI |
EC (assuming all pairs of Cys residues form cystine) |
EC (assuming all Cys residues are reduced) |
Half Life (hrs) |
II |
GRAVY |
-R |
+R |
AI |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
1 |
B. japonicum |
38383.33 |
353 |
9.30 |
18825 |
18450 |
20 |
45.32 |
-0.085 |
39 |
48 |
95.92 |
2 |
R. leguminosarum |
36926.28 |
343 |
8.96 |
10470 |
9970 |
20 |
30.65 |
-0.121 |
38 |
45 |
95.10 |
3 |
M. Ciceri |
38765.72 |
352 |
9.13 |
17460 |
16960 |
30 |
37.40 |
-0.261 |
43 |
52 |
90.43 |
Secondary Structure Predictions of Nif-A protein
To predict the secondary structural predictions of the nif A protein Chou-Fasman server19 and GOR20 was employed and the results were tabulated in table 3. The method implemented secondary structure predictions based on the analysis of relative frequencies of each amino acid in helices, sheets and turns anchored in the solved X-ray crystallographic protein template21.
Table (3):
Percentage of amino acids present in nif A protein estimated by UniProt software
S.No |
Amino acids |
B. japonicum (diazoefficiens) |
R. leguminosarum |
M.ciceri |
|---|---|---|---|---|
1 |
A (Ala) |
11.0% |
12.2% |
11.4% |
2 |
R (Arg) |
8.2% |
7.9% |
8.0% |
3 |
N (Asn) |
2.5% |
4.7% |
3.1% |
4 |
D (Asp) |
2.8% |
5.2% |
4.3% |
5 |
C (Cys) |
1.7% |
2.6% |
2.3% |
6 |
Q (Gln) |
2.5% |
3.8% |
4.0% |
7 |
E (Glu) |
8.2% |
5.8% |
8.0% |
8 |
G (Gly) |
6.5% |
8.7% |
6.8% |
9 |
H (His) |
0.8% |
1.2% |
1.4% |
10 |
I (Ile) |
4.8% |
5.5% |
5.1% |
11 |
L (Lue) |
11.0% |
11.4% |
10.5% |
12 |
K (Lys) |
5.4% |
5.2% |
6.8% |
13 |
M (Met) |
1.1% |
0.9% |
1.4% |
14 |
F (Phe) |
3.7% |
3.8% |
3.7% |
15 |
P (Pro) |
5.9% |
2.6% |
4.5% |
16 |
S (Ser) |
8.5% |
5.8% |
4.3% |
17 |
T (Thr) |
5.1% |
5.5% |
6.5% |
18 |
W (Trp) |
0.6% |
0.3% |
0.6% |
19 |
Y (Tyr) |
1.4% |
0.9% |
1.1% |
20 |
V (Val) |
7.9% |
5.5% |
6.2% |
Nif-A Protein Model Building and Evaluation
The linear amino acid sequence of Nif A protein of 3 different rhizobia retrieved from protein sequence database of uniprot (http://www.uniprot.org) 17 (The Accession No: for each organism was Q9AMY3 for B. japonicum, P09828 for R. leguminosarum and A0A165VD05 for M. ciceri). To produce the tertiary structures of proteins, templates were selected from PDB (Protein Data Bank)22 by using BLASTp algorithm23. Sequences of proteins that are more similar to the query sequence, were selected as templates. The modeling of the three dimensional structure of the proteins were performed by three homology modeling programs, Phrye224, Swissmodel25 and Modeller26. For the constructed 3D models energy minimization was performed to minimize steric collisions and strains without significantly altering the overall structure. Energy computations and minimization were carried out using the GROMOS96 force field27 and implementing Swiss-PDB Viewer. After optimization the 3D model were verified using the rampage28 and ProsA programs. PROSA web server is used to validate the modeled protein structure with available protein structure derived from PDB on the basic of z-score. Rampage server used for the validation of 3d structure modeled by plotting Ramachandran plot29, Solvent Accessible area etc.
Predicted primary protein sequence characterization of Nif A gene in B. japonicum, R. leguminosarum, M. ciceri
The nif A protein sequences of the selected rhizobia (B. japonicum, R. leguminosarum, and
M. Ciceri) were retrieved from the UniProt software 17. The details of the unique ID’s of Nif A for all the three species considered for further analysis are provided in table 1. UniProt is a universally acceptable database for the researchers to identify their specific protein’s knowledge regarding quality, richness, and accuracy with wide-range cross references and querying interfaces freely accessible30.
The primary structure was analyzed, and different parameters were computed using ExPasy ProtParam tool was tabulated in tables 2 and 331. The results suggested that the average molecular weight of Nif A proteins calculated is 38025.11 Da. Although the Expasy’s ProtParam computes the extinction coefficient for a range of (276, 278, 279, 280 and 282 nm) wavelength, 280 nm is favored, because proteins absorb strongly there while other substances commonly in protein solutions do not. The extinction coefficient of Nif A proteins at 280 nm was 18825, 10470, 17460 M-1cm-1 in B. japonicum, R. leguminosarum and M. Ciceri with respect to the concentration of Cys, Trp and Tyr (Table 3). The extinction coefficient of B. japonicum is comparatively high due to the high concentration of Tyr (1.4%). The computed protein concentration and extinction coefficients help in the quantitative study of protein-protein and protein-ligand interactions in solution32.
The instability index value for the Nif A proteins of B. japonicum, R. leguminosarum, and M. Ciceri were found to be 45.32, 30.65, 37.40, respectively. If instability index is below 40 then the protein is predicted as stable and above 40 it may be unstable33. Therefore nif A protein of R. leguminosarum, and M. Cicer is were found to be stable. The stable and compact condition of a protein (the pH at which the surface of the protein is charged while the net charge of the protein is Zero) is called the isoelectric point. The computed pI values of B. japonicum, R. leguminosarum, and M. Ciceri were 9.30, 8.96, 9.13 respectively which are more than 7, proving the alkaline nature of nif A protein. The computed isoelectric point (pI) will be useful for developing buffer systems for purification of the recombinant proteins by the isoelectric focusing method34. The total number of negatively charged residues (Asp + Glu) and total number of positively charged residues (Arg + Lys) are 39,48 in B. japonicum followed by 38, 45 in R. leguminosarum and 43, 52 in M. Ciceri respectively. Since the total negatively charged residues are comparatively lesser than the positively charged, it is understood that the protein is intercellular.
The half life of nif A protein sequence of B. japonicum, and R. leguminosarum was found to be 30 h with all the three domains where as it is 20 h in the absence of amino terminal domain. In M. Ciceri as the amino terminal is absent the half life is expected to be lesser, but interestingly it is found to be 30 h. Based on this prediction without amino terminal these two proteins were less stable.
The aliphatic index of a protein is defined as the relative volume occupied by aliphatic side chains, which include alanine, valine, isoleucine, and leucine, and contributes to protein thermostability35. The aliphatic index for the nitrogen fixing protein sequences were 95.92, 95.10, 90.43 for B. japonicum, R. leguminosarum and M. Ciceri respectively. The aliphatic index of nif A proteins results revealed that they are stable for a wide range of temperatures36. The Grand Average hydropathy (GRAVY) indices of nif A were -0.085, -0.121, -0.261 in B. japonicum, R. leguminosarum and M. Ciceri respectively. The Grand Average hydropathy (GRAVY) value for a peptide or protein is calculated as the sum of hydropathy values of all the amino acids, divided by the number of residues in the sequence37. This estimated low range values of nif A proteins were predicting that they are hydrophilic , possibility of better interaction with water.
All the protein polypeptide chains were prearranged with 20 amino acids. Each amino acid has its own characteristic to perform specific function of the protein. The percentage of polarity, charge, aliphatic and aromatic nature of proteins are vary based on their location and function. Phosphorylation is a vital procedure through which signaling pathways function. Three major amino acid residues namely Serine, Threonine and Tyrosine are mostly phosphorylated, as they contain hydroxyl group in their side chain and thus are capable of binding phosphate group38. All the 20 amino acids were estimated by using ProtoParm in which the highest percentage of amino acid is found in Alanine with 11.0, 12.2, 11.4 fallowed by Leucine with 11.0, 11.4, 10.5 and the lowest being typtophan with 0.6, 0.3, 0.6 in B. japonicum, R. leguminosarum, M.ciceri respectively (Table 3).
Prediction and characterization of Nif A secondary structures of B. japonicum, R. leguminosarum and M. ciceri
The prediction of the secondary structure of Nif A proteins were evaluated by using chou-Fasman method39 and GOR tools40. In our designed secondary structure of nif A protein, alpha helices were showing 43.6, 48.1, 47.44 percent in B. japonicum, R. leguminosarum, R. Ciceri respectively. It is followed by Random coils 41.36, 43.73, 41.76 and extended strands 15.58, 8.16, 10.80 (Table 4). Random coils have important functions in proteins for flexibility and conformational changes such as enzymatic turnover (reference). Our nif A protein revealed that the predominant nature of helix and coiling understood that the protein was more compact and strong bonded. As the globular structure and coiling nature of the protein assumed that our nif A protein is present in transmembrane region.
Table (4):
Prediction of secondary structure of nif A by Chou-Fasman method
| B. japonicum (diazo) | R. leguminosarum | R. ciceri | ||||
|---|---|---|---|---|---|---|
| Length | Percentage (%) | Length | Percentage (%) | Length | Percentage (%) | |
| Alpha helix (Hh) | 152 | 43.6 | 165 | 48.10 | 167 | 47.44 |
| 310 helix (Gg) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Pi helix (Ii) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Beta bridge (Bb) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Extended strand (Ee) | 55 | 15.58 | 18 | 8.16 | 38 | 10.80 |
| Beta turn (Tt) | 0.00 | 0.0 | 0.00 | 0.00 | 0.00 | 0.00 |
| Bend region (Ss) | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Random coil (Cc) | 146 | 41.36 | 150 | 43.73 | 147 | 41.76 |
| Ambiguous states | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
| Other states | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 |
In most of the nitrogen-fixing bacteria, the NifA protein binds to an upstream activating sequence (UAS) and acts in association with the RNA polymerase sigma factor RpoN (Ã54) to activate nif gene expression and, in rhizobia, the expression of several other symbiotic genes. The Nif A protein of B. japonicum and R. leguminosarum are composed of three domains: an amino (N)-terminal domain of unknown function, a central catalytic domain, and a carboxy (C)-terminal DNA-binding domain, but in M. ciceri amino (N)-terminal domain is absent. Between the central and the DNA binding domains, interdomain linker region was conserved in all these three rhizobial species. A few predictions have been made to find out the probable function of specific domains based on the comparison of amino acid sequence of Nif A proteins in three rhizobial species. A comparative low percentage of homology has been identified in the N-terminal region of B. japonicum and R. leguminosarum. A very high conservation in the sequence has been observed in the long central domain proposed to be responsible for the interaction with the RNA polymerase and/or with s54 (Figure 1). A region of considerable homology close to the C-terminus has been found, containing helix-turn-helix motif characteristic of DNA binding proteins.
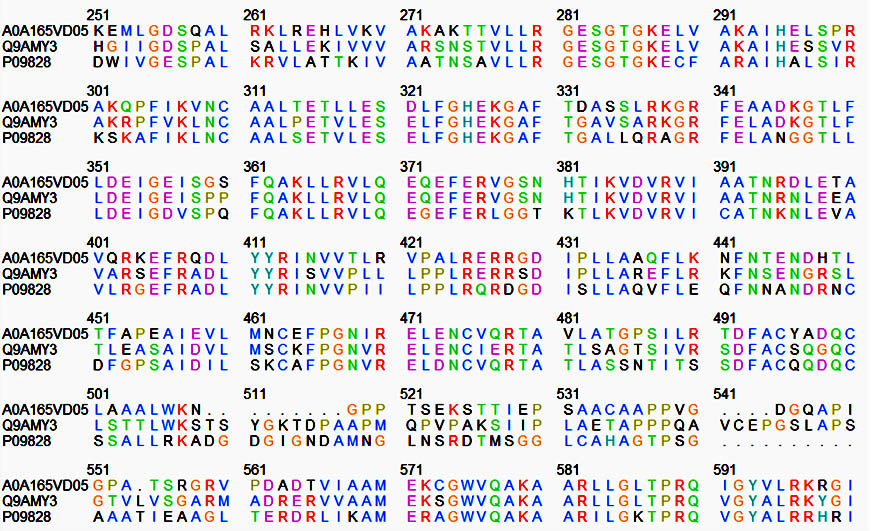
Fig. 1. Nif A protein sequence of Bradyrhizobium japonicum (Q9AMY3), Rhizobium leguminosarum (P09828), Mesorhizobium ciceri (P09828) by UniProt software
Between Phenylalanine-465 and Alanine-480 there are 15 identical amino acids in the three NifA sequences, five conserved cysteine residues at positions 310, 463, 475, 495 and 500. Role of the cysteines might be the binding of a cofactor (covalently bound heme or a complex [Fex:SX]- cluster) which is essential for Nif A activity of B. japonicum and R. leguminosarum. Proteins of this class also contain an additional invariant cysteine residue in the AAA+ domain. The presence of cysteine residues seems to correlate with the oxygen sensitivity of nif A proteins. This might suggest a model in which metal ion coordination to the cysteine residues control the activity of these proteins in response to the redox status.
3 D Modeling of Nif A Tertiary structure
There is a lack of experimental structures for nif A proteins considered. Out of the three domains of nif A protein, 3D structure was modeled for a central catalytic domain, and a carboxy (C)-terminal DNA-binding domain. The modeling of three dimensional structure of protein was performed by three homology modeling programs, Phyre2, Swiss and Modeller. The f and y distribution of the Ramachandran Map generated by non glycine, non proline residues were summarized in table 5. A comparison of the results obtained from the Phyre2, Swiss and Modeller, three different software tools were showed that the models generated by Modeller was more acceptable when compared to Phyre2 and Swiss Models.
Table (5):
Ramachandran plot calculation using rampage server
| Server | Ramachandran plot calculation | Bradyrhizobium japonicum | Rhizobium leguminosarum | Mesorhizobium ciceri |
|---|---|---|---|---|
| Phyre2 | Number of residues in favoured region | 85.3% | 92.1% | 93.4% |
| Number of residues in allowed region | 8.5% | 6.5% | 4.6% | |
| Number of residues in outlier region | 6.2% | 1.4% | 2.0% | |
| Swiss model | Number of residues in favoured region | 96.5% | 92.9% | 92.3% |
| Number of residues in allowed region | 3.1% | 6.3% | 6.5% | |
| Number of residues in outlier region | 0.4% | 0.8% | 1.1% | |
| Modeller | Number of residues in favoured region | 95.0% | 93% | 93.1% |
| Number of residues in allowed region | 3.0% | 3.7% | 3.7% | |
| Number of residues in outlier region | 2.0% | 3.4% | 3.1% |
Ramachandran plot calculation using rampage serverThe stereo chemical quality of the predicted models and accuracy of the protein model was evaluated after the refinement process using Ramachandran Map calculations computed with the Rampage program. The assessment of the predicted models generated by modeller was shown in figure 2a,b,c. The main chain parameters plotted are Ramachandran plot quality, peptide bond planarity, Bad non bonded interactions, main chain hydrogen bond energy, C-alpha chirality and over-all G factor. In the Ramachandran plot analysis, the residues were classified according to its regions in the quadrangle. The 3 Dimentional proteins designed for Nif A of B. japonicum, R. leguminosarum and M. ciceri were analyzed by modellar software and the results revealed that the allowed regions of residues are 96.8, 93, 93.1% respectively. The distribution of the main chain bond lengths and bond angles were found to be within the limits for these proteins. Such figures assigned by Ramachandran plot represent a good quality of the predicted models.
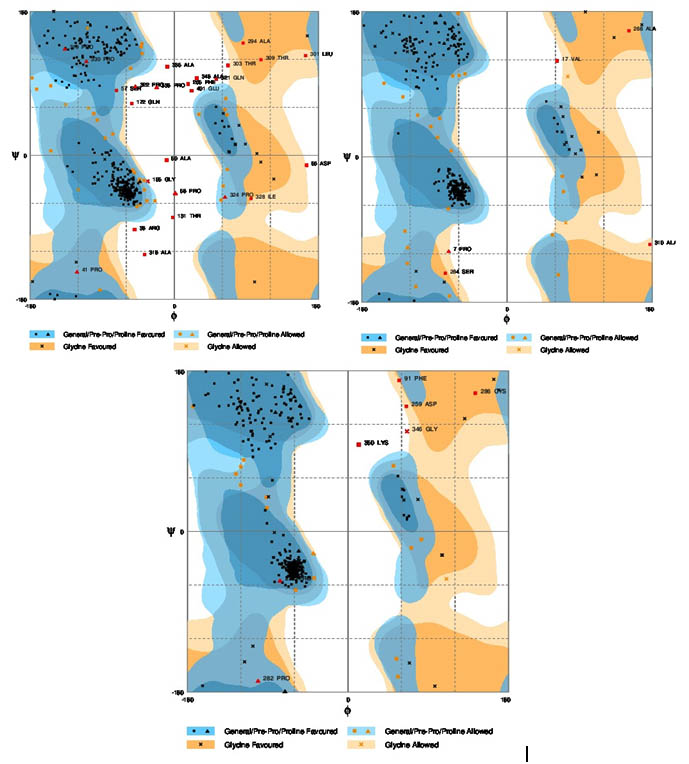
Fig. 2. Ramachandran plots for the three bacteria. a) Bradyrhizobium japonicum, b) Rhizobium leguminosarum, c) Mesorhizobium ciceri
The modeled structures of Nif A proteins were also validated by other structure verification servers, Prosa -web41. In the designed 3D protein, standard bond angles of the three models were determined using Prosa -web and the results were shown in table 6. The predicted structures conformed well to the stereochemistry indicating reasonably good quality. After model building, the structure was validated through energy minimization with Z-Score by using Prosa Web42. The Rampage-score provides an estimate of the absolute quality of a model by comparing it to same sized reference structures present in the PDB and solved by experimental techniques. Z-score was used to estimate the ‘degree of nativeness’ of the predicted structure. Z-score for modeled energy minimized PDB structure from Phyre2, Swiss and Modeller servers were -7.88, -7.43, -7.31, for B. japonicum, -8.26, -6.46, -7.18 for R. leguminosarum and -7.02, -6.18, -6.92 for M. ciceri respectively (Table 6). In this paper all the three i.e. Phyre2, Swiss and Modeller servers are showing similar values.
Table (6):
Z-scores for overall model quality using Prosa -web
Accession No |
Phyre2 |
Swiss model |
Modeller |
|---|---|---|---|
Q9AMY3 |
-7.88 |
-7.43 |
-7.31 |
P09828 |
-8.26 |
-6.46 |
-7.18 |
A0A165VD05 |
-7.02 |
-6.18 |
-6.92 |
To achieve optimistic results in biological nitrogen fixation a deep understanding of protein at structural level is essential. In silico studies provides an opportunity to accomplish the structural modeling and analysis of any protein. In the present study, Nif A sequences of B. japonicum, R. leguminosarum and M. ciceri were selected to determine the physicochemical properties and various protein structure levels using in silico techniques. Primary structure analysis revealed that most of the Nif A employed in the current study was hydrophilic in nature and presence of cysteine residues seems to correlate with the oxygen sensitivity of these proteins. The secondary structure analysis confirmed that in most of the sequences, alpha helix dominated followed by an random coil, extended strand and beta turns. Tertiary structure predictions were analyzed by three different homology servers Phyre 2, Swiss model and Modeller. The models were validated by protein structure checking tool called Rampage. Out of three servers our results revealed that the Modellar is acceptable in silico tool for the designed Nif A protein. We hope that our future studies with the quaternary structure of Nif A protein will provide a better incite of exact or most probable molecular mechanisms involved in nitrogen fixation in the present three rhizobial strains. One of the challenging research goals in the future is to elucidate the mechanism whereby the Nif A protein ultimately responds to the redox state in the cell.
ACKNOWLEDGMENTS
No public or private financial support has been taken for publishing this project. The project was conducted on self finance mode. Authors thank Dr. K. Srinivasulu, Head, Dept of Biotechnology, KL University for his support in research. Express gratitude to Dr. Vijaya Saradhi for his moral support and inspiration.
- Gergely Maróti, ÉvaKondorosi. Nitrogen-fixingRhizobium-legume symbiosis: are polyploidy and host peptide-governed symbiont differentiation general principles of endosymbiosis? Front. Microbiol. 2014; 5 . doi: 10.3389/fmicb.2014.00326.
- Gopalakrishnan S, Sathya A, Vijayabharathi R, Varshney RK, Gowda CLL, Krishnamurthy L. Plant growth promoting rhizobia: challenges and opportunities. 3 Biotech. 2015; 5: 355-377. https://link.springer.com/article/10.1007/s13205-014-0241-x.
- Hill S, Austin S, Eydmann T, Jones T, Dixon R. Azotobacter vinelandii NIFL is a flavoprotein that modulates transcriptional activation of nitrogen-fixation genes via a redox-sensitive switch. Proc Natl Acad Sci U S A.; 1996; 93(5): 2143-8. https://www.ncbi.nlm.nih.gov/pubmed/8700899.
- Black M, Moolhuijzen P, Chapman B, Barrero R, Howieson J, Hungria M, et al. The genetics of symbiotic nitrogen fixation: comparative genomics of 14 rhizobia strains by resolution of protein clusters. Genes (Basel). 2012; 3(1):138-66. doi: 10.3390/genes3010138. https://www.ncbi.nlm.nih.gov/pubmed/24704847.
- Fischer H. Genetic Regulation of Nitrogen Fixation in Rhizobia. Microbiological Reviews. 1994; 58(3): 352-386. https://www.ncbi.nlm.nih.gov/pubmed/7968919.
- Passaglia LM, Van Soom C, Schrank A, Schrank IS. Purification and binding analysis of the nitrogen fixation regulatory NifA protein from Azospirillum brasilense. Braz J Med Biol Res.; 1998; 31(11):1363-74. https://www.ncbi.nlm.nih.gov/pubmed/9921270.
- Macheroux P, Hill S, Austin S, Eydmann T, Jones T, Kim SO. Electron donation to the flavoprotein NifL, a redox-sensing transcriptional regulator. Biochem J. 1998; 332 ( Pt 2): 413-9. https://www.ncbi.nlm.nih.gov/pubmed/9601070.
- Zehr JP, Kudela RM. Nitrogen cycle of the open ocean: from genes to ecosystems. Ann Rev Mar Sci. 2011; 3:197-225. https://www.ncbi.nlm.nih.gov/pubmed/21329204.
- Little R, Colombo V, Leech A, Dixon R. Direct interaction of the NifL regulatory protein with the GlnK signal transducer enables the Azotobacter vinelandii NifLNifA regulatory system to respond to conditions replete for nitrogen. J Biol Chem.; 2002; 277: 15472–15481. http://www.jbc.org/content/277/18/15472.full.
- Satyanarayana SDV, Krishna MSR, Pindi PK. A Strategic Approach for Isolation and Identification of Plant Growth Promoting Rhizobial Strains from Bhadrachalam Forest Area with Respect to Groundnut Cultivar. Ind J Sci Tec.; 2017; 10(5). doi: 10.17485/ijst/2017/v10i5/105214. http://www.indjst.org/index.php/indjst/article/view/105214.
- Gupta CL, Akhtar S, Bajpai P. In silico protein modeling: possibilities and limitations. EXCLI J.; 2014; 13: 513–515. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4467082/.
- Fiser A. Template-Based Protein Structure Modeling. Methods Mol Biol. 2010; 673: 73–94. doi: 10.1007/978-1-60761-842-3_6. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4108304/.
- Berger DK, Narberhaus F, Lee HS, Kustu S. In vitro studies of the domains of the nitrogen fixation regulatory protein NIFA. J Bacteriol., 1995; 177(1):191-9. https://www.ncbi.nlm.nih.gov/pubmed/8002617.
- Huala E, Ausubel FM. The central domain of Rhizobium meliloti NifA is sufficient to activate transcription from the R. meliloti nif H promoter. J Bacteriol.; 1989; 171(6):3354-65. https://www.ncbi.nlm.nih.gov/pubmed/2722751.
- Morett E, Buck M. NifA-dependent in vivo protection demonstrates that the upstream activator sequence of nif promoters is a protein binding site. Proc Natl Acad Sci U S A.; 1988; 85(24):9401-5. https://www.ncbi.nlm.nih.gov/pubmed/2849102.
- Morett E, Buck M. In vivo studies on the interaction of RNA polymerase-sigma 54 with the Klebsiella pneumoniae and Rhizobium meliloti nifH promoters. The role of NifA in the formation of an open promoter complex. J Mol Biol.;1989; 210(1):65-77. https://www.ncbi.nlm.nih.gov/pubmed/2685331.
- UniProt Knowledgebase tool. Available from: http://www.uniprot.org/.
- ProtParam tool. Available from: http://web.expasy.org/protparam/.
- CFSSP: Chou & Fasman Secondary Structure Prediction Server. Available from: http://www.biogem.org/tool/chou-fasman/.
- Garnier J, Gibrat JF, Robson B. GOR method for predicting protein secondary structure from amino acid sequence. Methods Enzymol.; 1996; 266:540-53. https://www.ncbi.nlm.nih.gov/pubmed/8743705.
- Kumar TA. CFSSP: Chou and Fasman Secondary Structure Prediction server. Wide Spectrum. 2013; 1(9): 15-19. https://sandbox.zenodo.org/record/50733#.WfQqd1v-nce.
- Bernstein FC, Koetzle TF, Williams GJ, Meyer EF Jr, Brice MD, Rodgers JR, et al. The Protein Data Bank. A computer-based archival file for macromolecular structures. Eur J Biochem.; 1977; 80(2):319-24. https://www.ncbi.nlm.nih.gov/pubmed/923582.
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol.; 1990; 215(3):403-10. https://www.ncbi.nlm.nih.gov/pubmed/2231712.
- Kelley A, Mezulis S, Yates CM, Wass NM, Sternberg MJE. The Phyre2 web portal for protein modeling, prediction and analysis. Nature Protocols, 2015; 10, 845–858. doi:10.1038/nprot.2015.053. http://www.nature.com/nprot/journal/v10/n6/full/nprot.2015.053.html.
- Arnold K, Bordoli L, Kopp J, Schwede T. The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 2006; 22(2):195-201. https://www.ncbi.nlm.nih.gov/pubmed/16301204.
- Sali A, Blundell TL. Comparative protein modelling by satisfaction of spatial restraints. J Mol Biol. 1993; 234(3):779-815. https://www.ncbi.nlm.nih.gov/pubmed/8254673.
- Scott WRP, Hunenberger PH, Tironi IG, Mark AE, Billeter SR, Fennen J, et al. The GROMOS Biomolecular Simulation Program Package. J. Phys. Chem. A. 1999; 103 (19):3596–3607. http://pubs.acs.org/doi/abs/10.1021/jp984217f.
- Rampage: Ramachandran Plot Analysis. Available from: http://mordred.bioc.cam.ac.uk/~rapper/rampage.php.
- Ramachandran GN, Ramakrishnan C, Sasisekharan V. Stereochemistry of polypeptide chain configurations. J Mol Biol. 1963; 7: 95-9. https://www.ncbi.nlm.nih.gov/pubmed/13990617.
- Apweiler R, Bairoch A, Wu CH, Barker WC, Boeckmann B, Ferro S. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 2004; 32(Database issue): D115–D119. doi: 10.1093/nar/gkh131. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC308865/.
- Expays protoparam tool for protein primary structure analysis. Available from: https://web.expasy.org/protparam/.
- Gill SC, von Hippel PH. Calculation of protein extinction coefficients from amino acid sequence data. Anal Biochem. 1989; 182(2):319-26. https://www.ncbi.nlm.nih.gov/pubmed/2610349.
- Idicula-Thomas S, Balaji PV. Understanding the relationship between the primary structure of proteins and its propensity to be soluble on overexpression in Escherichia coli. Protein Sci. 2005; 14(3): 582–592. doi: 10.1110/ps.041009005. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2279285/.
- Adhikari S, Manthena PV, Sajwan K, Kota KK, Rabindra R. A unified method for purification of basic proteins. Anal Biochem. 2010; 400(2): 203–206. doi: 10.1016/j.ab.2010.01.011. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4231783/.
- Ikai A. Thermostability and aliphatic index of globular proteins. J Biochem. 1980; 88(6): 1895-8. https://www.ncbi.nlm.nih.gov/pubmed/7462208.
- Sivakumar K, Balaji S, Radhakrishnan G. In silico characterization of antifreeze proteins using computational tools and servers. J. Chem. Sci. 2007; 119(5): 571–579.
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. J Mol Biol. 1982; 157(1):105-32. https://www.ncbi.nlm.nih.gov/pubmed/7108955.
- Understanding Amino Acid Side Chain Characteristics for the MCAT. 2015. Available from: http://leah4sci.com/understanding-amino-acid-side-chain-characteristics-for-the-mcat/.
- Chen H, Gu F, Huang Z. Improved Chou-Fasman method for protein secondary structure prediction. BMC Bioinformatics. 2006; 7(Suppl 4): S14. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1780123/.
- Sen TZ, Jernigan RL, Garnier J, Kloczkowski A. GOR V server for protein secondary structure prediction. Bioinformatics. 2005; 21(11):2787-8. https://www.ncbi.nlm.nih.gov/pubmed/15797907.
- Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007; 35(Web Server issue): W407–W410. doi: 10.1093/nar/gkm290. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1933241/.
- Goswami AM. Structural modeling and in silico analysis of non-synonymous single nucleotide polymorphisms of human 3²-hydroxysteroid dehydrogenase type 2. Meta Gen. 2015; 5: 162-172. doi: 10.1016/j.mgene.2015.07.007. https://www.ncbi.nlm.nih.gov/pubmed/26288759.
© The Author(s) 2018. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.