


ISSN: 0973-7510
E-ISSN: 2581-690X
Nipah virus is a pleomorphic virus that causes high mortality with unpredictable outbreaks. The virus also shows high zoonotic potential with long term neurological damage after recovery further adding to the disease burden. An in-silico epitope-based vaccine offers a promising solution to supplement wider efforts to control the viral spread. This is achieved through immunoinformatics approach using a plethora of servers available. We derived cytotoxic T-cell, T-Helper, B-cell and IFN-γ targeting epitopes from surface glycoprotein G. Cytotoxic T-cell specific epitopes, HLA-B*4402, chimeric multiepitope vaccine structures were prepared using homology modelling method. The structures were validated using various methods and docking simulation was performed between epitopes and HLA-B*4402. Similarly, the vaccine construct was docked to Toll like receptor-4 and a molecular dynamics simulation was performed to assess stability of interaction. Both the docking simulations showed stable interactions with their respective receptors. Immune-simulation was carried out to validate the efficacy of vaccine candidate which showed elevated levels of antibodies such as IgM and IgG due to increase in active B cell population. Both in-vitro and in-vivo serological analysis is required for confirmation of vaccine potency. To facilitate this effort, codon optimization was undertaken to remove existing codon bias. The optimized gene sequence was cloned into the PUC19 vector to express in Escherichia coli K12 strain. Additionally, a poly histidine (6xHis) tag was added at the C-terminal end to ease the purification step. The immune-informatics approach hopes to accelerate vaccine development process to reduce the risk of attenuation while increasing the success rates of pre-clinical trials.
Nipah virus, vaccine, immune-simulation, immunoinformatics, molecular dynamics, cytokine
Nipah virus (NiV) is a highly pathogenic virus belonging to the family Paramyxoviridae and order Mononegavirals1. NiV infection were first reported in Malaysia in 1998 associated with encephalitis and a high mortality rate of 39.6%2. Subsequent outbreaks in 2001 emerged in the Indian subcontinent demonstrating an increase in fatality rates as high as 70% in some regions3. Fruits bats (Pteropus) are known to be the natural reservoir of the virus, with pigs (Sus scrofa) reported to be the intermediate hosts in some outbreaks1-5. Interestingly, Hendra virus (HeV) which was shown to infect horses was observed to be genetically related to the Nipah virus with protein-coding regions bearing a similarity of 68-92% and hence grouped into the genus Henipavirus 5. Both the viruses demonstrate broad host-specificity, a trait rarely associated with other members of Paramyxoviridae 5. Recent outbreak of NiV was reported in Kerala, India with 18 confirmed cases and 17 fatalities showing a mortality rate of 94.4% NiV is pleomorphic and constitutes an 18.2 Kb negative-sense single stranded RNA based genome 7. Sequencing and characterization of NiV genome was carried out successfully in many bat species across Asia 8. They include Pteropus hypomelanus, Pteropus lylei, Pteropus medius and Pteropus vapmyrus 8. Studies revealed the presence of non-coding RNA sequence specifically a 3′ Untranslated region (UTR) occupying major portion of the viral genome. This is responsible for longer genomes in genus Henipaviruses compared to other members of the Paramyxoviridae family (~15 Kb genome length) 9. The function of the 3′ UTR is to perform effective post-transcriptional utilization of cell machinery during viral replication9. The NiV proteome contains nine important proteins including six structural and three non-structural proteins 10. The nucleoprotein (N) is responsible for viral replication and genome wrapping, Matrix protein (M) for structural arrangement and release, Large polymerase (L), Phosphoprotein (P) along with non-structural accessory proteins namely V-protein, C-protein and W-protein play an important role in suppressing α/β interferon (IFN-α/β) production thereby evading innate immune response 10,11. This is achieved by the downregulation of JAK-STAT pathway wherein these proteins competitively bind to STAT-1 and inhibit its phosphorylation in the presence of IFN-α/β) 11. The viral structure also includes two surface glycoprotein namely G protein and F protein 12. The G protein is 602 amino acids long primarily responsible for host cell adhesion 12. It interacts with Ephrin B2 and B3 receptors of the host cell leading to virion internalization by means of clathrin-mediated endocytosis 12,13. The F protein has 546 amino acids and causes viral-host cell fusion forming a multinucleated syncytium for viral replication initiation13. NiV primarily infects the Central Nervous System (CNS) via cranial nerves. Incubation period ranges between 4 to 60 days. Infected patients are diagnosed with the presence of viral loads in the Cerebrospinal fluid (CSF) and tend to suffer from febrile-encephalitis including thrombosis, parenchymal necrosis and vasculitis 14. Occasionally, vasculitic lesions are also found in respiratory tract, kidneys and heart14,15. The in- silico immunoinformatics approach offers a promising solution towards novel vaccine design. The method involves identification of antigenic epitopes within antigenic proteins which can be used to elicit both cellular and humoral immune responses16. It works by the specific interaction between the isolated antigenic epitopes with Major Histocompatibility Complex (MHC) and Human Leukocyte Antigen (HLA) molecules during antigen presentation17. Epitope based vaccines eliminate the risk associated with attenuation and adds the benefit of an in–silico validation prior to in vitro trials16,17. It also reduces time and cost associated with the vaccine development. Surface glycoproteins such as G protein and F protein are considered ideal targets for the immune system due to their interaction with the host cell receptors and exposure on the viral surface 12. The immunoinformatics approach hopes to derive epitopes from the surface glycoproteins particularly the G and F protein and validate the immune response in silico using immune system simulation.
Identification of glycoprotein antigenicity
The surface glycoproteins G-protein (Uniprot ID: Q9IH62) and F-protein (Uniprot ID: Q9IH63) FASTA sequences were screened for antigenicity using Vaxigen 2.0 server 18with the most probable protective antigens being predicted rather than requiring causative microorganisms to be grown. Despite the obvious advantages of this approach – such as speed and cost efficiency – its success remains dependent on the accuracy of antigen prediction. Most approaches use sequence alignment to identify antigens. This is problematic for several reasons. Some proteins lack obvious sequence similarity, although they may share similar structures and biological properties. The antigenicity of a sequence may be encoded in a subtle and recondite manner not amendable to direct identification by sequence alignment. The discovery of truly novel antigens will be frustrated by their lack of similarity to antigens of known provenance. To overcome the limitations of alignment-dependent methods, we propose a new alignment-free approach for antigen prediction, which is based on auto cross covariance (ACC. The threshold was set at 0.4 while the target organism being selected for was a Virus. The selection threshold was determined to be >0.5. The workflow for the in-silico methodology can be found in [Fig. 1].
Identification of T-cytotoxic cell targeted epitopes
T-lymphocytes are critical in mounting an immune response against invading pathogens. T-cell epitopes were derived from the antigenic G-protein using NetCTL1.2 server19. The threshold was selected to be 0.5 while sensitivity and specificity were 0.89 and 0.94 respectively. The epitopes were Major Histocompatibility complex (MHC) supertype A1 specific and the length was adjusted to the maximum allowed 9 aminoacid length residues to improve viral sequence recognition by the immune system. Potential peptides were obtained from which MHC specific ligands were simultaneously isolated. The potential MHC specific epitopes were then screened using Immune Epitope Database and Analysis resource (IEDB) T-cell epitope prediction tool20. The epitopes were particularly screened against antiviral MHC-1 supertype alleles such Human leukocyte antigens HLA-B*44, HLA-B*35, HLA-B*32 and HLA-A*1 using stabilized matrix base method21. The identified antigens were subjected to MHC-1 immunogenicity scoring in IEDB. The selection criteria were a positive score on MHC-1 antigenicity.
Design and validation of three-dimensional structures of epitopes and HLA-B*4402
The three-dimensional structures of epitopes were prepared using PEPFOLD3 server in Protein data bank (PDB format) 22. The lowest energy models were selected for dock preparation. The structure of HLA-B*4402 was prepared based on homology modelling method using I-TASSER server using PDB ID:1M6O as a template23. HLA-B*4402 was adopted due to its role in imparting anti-viral protective effects21. The lowest energy model was chosen and subjected to structure refinement employing ModRefiner server24. The final refined model was further validated using ERRAT2, verify 3D, QMEAN, PROVE and RAMPAGE servers25-27.
Molecular Docking and analysis of T-cell specific epitopes with HLA-B*4402
Ligand binding site in HLA-B*4402 was identified using neural network based method in DeepSite server28. The structures for docking were prepared using Dockprep tool in UCSF chimera. Solvent and un-complexed ions were removed. Charges and missing hydrogens were added back. Charge allocation was carried out using AMBER ff14SB force fields. The grid size (X, Y and Z) were 88.4709, 71.5214 and 79.2498 respectively. Molecular docking was performed using AutoDock Vina on UCSF chimera29,30. The binding region was within the ligand binding site identified earlier. Binding affinity values for all the epitopes were obtained in negative values of Kcal/mol. The Epitope-HLA-B*4402 complexes were further used to analyze dissociation constant (KD) to assess the stability of the complex. The lower the KD value stronger the interaction. Binding free energy was measured using Molecular Mechanics Born Solvent Accessibility (MMGBSA) approach. Dissociation constant value for each of the complexes were predicted using PRODIGY server31, MMGBSA binding free energy of the complex values were obtained using HAWKDOCK server32.
Prediction of T-Helper cell (CTH) targeted epitopes
T-Helper cell epitopes were derived from G-protein using NetMHC-II 2.3 server 33. The threshold for strong binders was selected to be (2% rank) while the threshold for weak binders was set at (10% rank). The peptide length was adjusted to 15 residues in default mode.
Prediction of B-cell linear epitopes
B-cell linear epitopes were derived using BepiPred Linear epitope prediction 2.0 method on IEDB servers34 and SVMTriP servers35. While the length of the epitopes in SVMTriP was set to be 14 residues long, the length remained variable in the BepiPred Linear epitope prediction 2.0 method. B-cell linear (continuous) epitopes were predicted as they contiguous with the protein sequence from which they are derived as opposed to the discontinuous B-cell epitopes66.
Prediction of Interferon-γ (IFN-γ) targeted epitopes
Interferon-gamma plays a crucial role in antiviral response, it is important to derive epitopes activating this critical interleukin. IFN-γ specific epitopes were screened from G-protein using IFNepitope server36. The peptide length was adjusted to be 15 residues long. The prediction approach used both motif and SVM based while the model was selected to be IFN-g vs non IFN-g. The method used was called MERCI algorithm36.
Design of Multi-epitope vaccine candidate
A multi-epitope vaccine constituting different types of epitopes was constructed. CTL specific epitopes for immunogenicity, HTL targeted epitopes for cytokine eliciting characteristics, B-cell specific epitopes and IFN-γ for interferon response were selected. Toll like receptor (TLR-4) agonist (Uniprot: P9WHE3, 50S ribosomal protein L7/L12 OS=Mycobacterium tuberculosis [strain ATCC 25618 / H37Rv]) was used as an adjuvant at the N-terminal37. EAAAK linker was used to connect the adjuvant to the CTL specific epitope38, while subsequent CTL specific epitopes were linked using AAY linkers38. Here the CTL epitope FLVRTEFKY was excluded as it shares the aminoacid residues common in the epitope VGFLVRTEF from same position. T-Helper cell targeted epitopes were joined to the former using GPGPG linkers38 while B-cell linear epitopes and an IFN-γ epitope were associated with the help of KK linkers38. A poly histidine (6xHis) tag was added at the C-terminal for ease of purification39. The In-silico 3D structure of the multi-component vaccine was prepared in PDB format using Phyre 2 server based on homology modelling40 for assessment of physiochemical properties and perform molecular docking simulation with TLR-4. The model with the lowest energy was identified and subjected towards refinement using 3Drefine and Galaxy refine servers41,42. The final model was validated using RAMPAGE, ProSA, ERRAT2 and QMEAN servers 25–27,43,44.
Assessment of Antigenicity, Allergencity and Toxic properties of the Multi-epitope vaccine candidate
The antigenicity of the vaccine candidate was estimated using ANTIGENpro server45. Allergenicity of the vaccine was predicted using AllergenFP 1.0 and AllerTop 2.0 servers46,47. The toxicity of the vaccine was determined using ToxinPred server 48.
Assessment of Physiochemical properties and secondary structure
Physiochemical properties of the multi-epitope protein were determined using ProtParam and SOLpro servers49,50. Parameters such as theoretical isoelectric point, molecular weight, extinction coefficient, solubility upon overexpression, half-life, instability index, grand average of hydropathicity (GRAVY), aliphatic index were assessed. Secondary structure bearing regions were identified employing PSPIRED, RaptorX and BETApro servers 51,52.
Molecular Docking of Multi-epitope vaccine candidate with Toll like Receptor 4
To validate the function of the adjuvant, Molecular docking was performed between the multi-epitope vaccine and TLR-4 (PDB ID:4G8A) using rigid body docking method on ClusPro 2.0 docking server53. The option of graphical processing unit (GPU) based server was chosen to improve accuracy. The model with the lowest energy was selected and analyzed. The protein-protein interaction regions were assessed using an interaction map generated with the help of CoCoMaps server54. The interacting residues of the complex were subjected to HotRegion server analysis to identify Hotspots within the interactions55. Further, the binding affinity (∆G) and Dissociation constant (KD) were analyzed using PRODIGY server31 and the free energy of the complex was predicted using MMGBSA method employing HAWKDOCK server32.
MD simulation of the multi-epitope vaccine-TLR-4 complex
Normal mode analysis (NMA) was executed on IMODS server to study protein structure stabilization and large-scale mobility. Eigen values were obtained to determine the molecular stiffness56. Elastic network analysis of the complex and mobility was derived using DynOmics 1.0 server 57.
Immune-simulation of the multi-epitope vaccine candidate
Immuno-simulation was performed using C-IMMSIM server57. This is an agent based in silico technique which uses position-specific scoring matrix (PSSM) and machine learning algorithms to simulate immune response to the vaccine injection57. To test the efficacy of the vaccine a total of 3 doses were injected at a 4-week interval. Time steps of 1,84 and 168 were chosen for second and third boosters with each time-step equal to 8 hours hence 300 simulation steps. The vaccine was not coated with Lipopolysaccharide and the alleles HLA-A*32:15, HLA-A*01:01, HLA-B*35:01 and HLA-B*4402 alleles specific activity was selected. The simulation results were further analyzed to assess the immune response.
Codon optimization and In-silico cloning
To express the Nipah Virus G-protein based vaccine construct in Escherichia coli K12 strain, codon optimization was performed to eliminate the risk of codon bias. This was achieved employing Java codon Adaptation tool (JCat)58. Additionally, three more parameters were selected to prevent the use of rho independent transcription termination, prokaryotic ribosome binding site and cleavage site for certain restriction enzymes59. The optimized gene sequence obtained was then used to clone the gene in PUC19 vector with the help of SnapGene tool59.
Highest antigenic Glycoprotein identification
The surface G-protein (Uniprot ID: Q9IH62) showed antigenicity score of 0.51 (>0.5) while the fusion F-protein (Uniprot ID: Q9IH63) gave a score 0.47 (<0.5). The minimum threshold to select antigenic protein was set at the score of 0.5. Hence G-protein was selected to derive antigenic epitopes for vaccine construction.
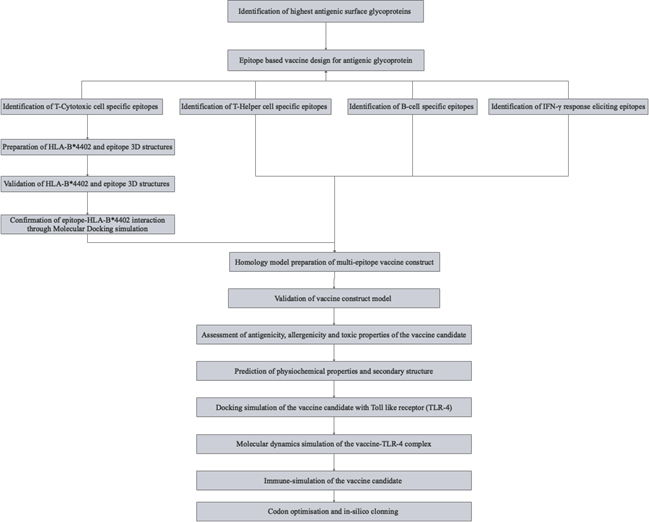
Fig. 1. Schematics of the methodology adopted in designing the vaccine candidate.
Identification of T-cell epitopes
Using the NetCTL1.2 server, a total of 590 potential peptides were obtained from which 52 were identified as being MHC specific ligands. The 52 potential epitopes were further screened using Immune Epitope Database and Analysis resource (IEDB) T-cell epitope prediction tool. The epitopes were particularly screened against antiviral MHC-1 supertype alleles such Human leukocyte antigens HLA-B*44, HLA-B*35, HLA-B*32 and HLA-A*1. Five epitopes were derived based on the Half maximal inhibitory concentration (IC50>200nM). These five antigens were subjected to MHC-1 immunogenicity scoring in IEDB. All the five epitopes demonstrated positive scores [Table 1] indicating a promising role in T-cytotoxic cell specific antigenicity.
Table (1):
MHC-I immunogenicity scores for G-protein based epitopes.
Sl. No |
Position |
Epitope |
Immunogenicity score |
|---|---|---|---|
1. |
500-508 |
PEICWEGVY |
0.35162 |
2. |
223-231 |
AMDEGYFAY |
0.25913 |
3. |
367-375 |
VGFLVRTEF |
0.20702 |
4. |
532-540 |
AENPVFTVF |
0.19402 |
5. |
369-377 |
FLVRTEFKY |
0.17033 |
Design and validation of three-dimensional structures of epitopes and HLA-B*4402
The 3D structure of HLA-B*4402 was constructed using homology modelling. The prepared structure was then validated using RAMPAGE [Fig. 2]. Ramachandran plot showed 265 (96.7%) of the residues were in the favored region while 7 (2.6%) were in the allowed region only 2 (0.7%) residues were found in the outlier region [Fig. 2]. The ERRAT2 analysis showed an overall quality factor of 90.299 wherein a score of >80 is needed for an ideal structure. Further, VERIFY3D validation showed 98.91% of the residues to be scoring >=0.2 score with a ‘PASS’ designation. To pass the model must show a minimum >=0.2 score for >80% of the residues in the 3D model. The QMEAN showed -0.95 with overall score of 0.83 where the value lies in between 0 to 1 indicating a good model [Fig. 2].
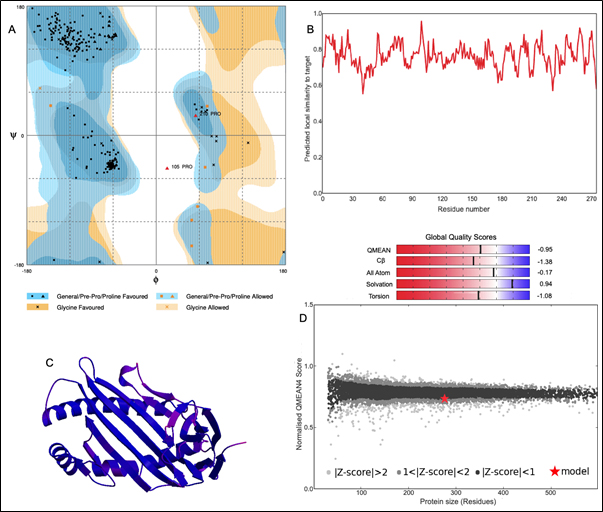
Fig. 2. Structural validation by (A) RAMPAGE derived Ramachandran plot, (B) Local quality estimate with QMEAN4, (C) PDB model of HLA-B*4402, (D) Comparison with non-redundant set of PDB structures.
Molecular Docking of T-cell epitopes with HLA-B*4402
Ligand binding site was identified using DeepSite server. The docking of the epitope AMDEGYFAY showed the highest binding affinity of -8.2 Kcal/mol using AutoDock Vina. However, the epitope PEICWEGVY-HLA-B*4402 complex demonstrated superior binding free energy (MMGBSA) of -47.66 Kcal/mol. Overall, the epitope AENPVFTVF binding indicated a stable interaction with -6.9 Kcal/mol [Table 2, Fig. 3] including a KD of 2.8×10-7 and -33.23 Kcal/mol binding free energy of the complex using HAWKDOCK [Table 2]. The docking results for all the epitopes are shown in the [Fig. 3]. As covalent bonding is rare among protein-protein interactions, hydrogen bonding is a stable form of non-covalent interactions taking place and enhances binding affinity depending on the inter-molecular distance (Å)66. There were three hydrogen bonding interactions formed wherein ASN 3 of AENPVFTVF interacted with TYR 27 of HLA-B*4402 with a bond length of 2.79 Å while other hydrogen bonding interactions between THR 7 and ASP 30, GLU 2 and ARG 48 respectively [supplementary Table 1]. The epitope PEICWEGVY formed the seven hydrogen bonds with HLA-B*4402 while the epitope AMDEGYFAY formed five hydrogen bonding interactions [Supplementary Table 1].
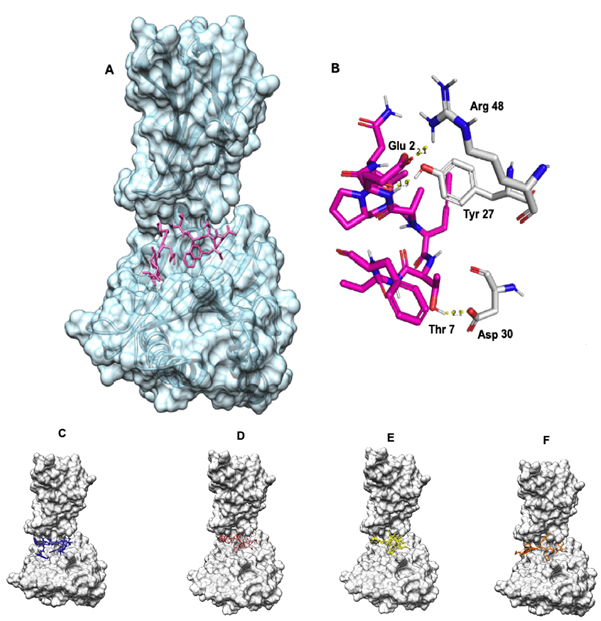
Fig. 3. (A) AENPVFTVF-HLA-B*4402 complex where the epitope is coloured in pink (B) Hydrogen bonding where the epitope is coloured in pink while the MHC molecule is coloured in white. Blue represents nitrogen and red oxygen. (C) FLVRTEFKY-HLA-B*4402 complex where the epitope is coloured in blue. (D) PEICWEGVY-HLA-B*4402 complex where the epitope is coloured in brown. (E) VGFLVRTEF-HLA-B*4402 complex where the epitope is coloured yellow. (F)AMDEGYFAY-HLA-B*4402 complex where the epitope is coloured in orange.
Table (2):
T-Cytotoxic cell specific epitope-HLA-B*4402 docking simulation results.
Sl. No |
Epitope |
Binding affinity (Kcal/mol) |
Binding Free energy of the complex (Kcal/mol) |
Dissociation constant (KD) |
|---|---|---|---|---|
1. |
PEICWEGVY |
-7.6 |
-47.66 |
4.2×10-7 |
2. |
AMDEGYFAY |
-8.2 |
-33.49 |
2.8X10-7 |
3. |
VGFLVRTEF |
-6.6 |
-33.20 |
1.0X10-6 |
4. |
AENPVFTVF |
-6.9 |
-33.23 |
4.7X10-7 |
5. |
FLVRTEFKY |
-6.6 |
-35.89 |
1.1X10-6 |
Identification of T-Helper specific, B-cell specific and IFN-γ epitopes
A total of 584 T-Helper specific epitopes were obtained of which 13 epitopes are considered as strong binders. Three epitopes were chosen to be used in the construction of chimeric multi-epitope vaccine they are QGDTLYFPAVGFLVRT, PANIGLLGSKISQST and DSKILSAFNTVIALL. Similarly, B-specific epitopes were identified by both IEDB and SVMTriP servers. Interestingly, one epitope with a maximum score of 1.0 was recommended by SVMTriP server while a list of B-cell epitopes was recommended by BepiPred Linear epitope prediction 2.0 method without a score. Three B-cell specific epitopes were selected to be used in the vaccine. These include, IALLGSIVIIVMNI, RSTDNQAV and SCSRGVS. A total of 88 IFN-γ stimulating epitopes were identified using IFNepitope server. However, we found many high scoring IFN-γ specific epitopes to be already part of the sequences forming the B-cell and T-helper specific epitopes. Therefore, two epitopes which did not form part of the B-cell or T-cell specificity with positive scores were chosen to be included in the chimeric multi-epitope protein molecule. They are LVVNWRNNTVISRPG and PLVVNWRNNTVISRP with scores 0.6239, 0.5711 respectively. Here a repetition of the similar sequence with additional amino acids was done to stimulate maximum IFN-γ response as observed in [Fig. 11F].
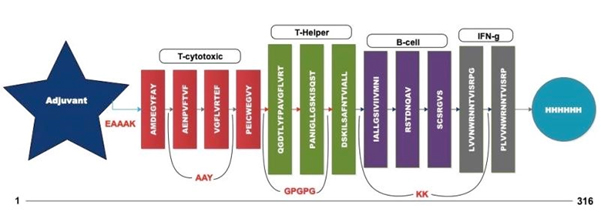
Fig. 4. Multi-epitope vaccine construct outline
Multi-epitope vaccine construct with Antigenicity, Allergencity and Toxicity prediction
A chimeric multiepitope vaccine candidate with 316 amino acids was constructed using homology modelling in Phyre2. As shown in [Fig. 4], the adjuvant was added at the N-terminal end while a His-tag was added at the C-terminal for the ease of purification. The linkers EAAAK, AAY, GPGPG and KK linkers were used in connecting the specific epitopes. The initial model obtained in the PDB format was subjected to the model refinement using 3Drefine. The model with the 3Drefine score of 30372.8 with MolProbity score of 3.799 was selected. Further refinement using Galaxy refine improved the MolProbity score to 2.704 and the model rama-favored with 87.6%. This 3D structure under RAMPAGE investigation showed 281 (89.5%) in favored region while 21 (6.7%) in the allowed region. 12 (3.8%) residues were in the outlier region. Additionally, ERRAT2 showed model quality of 80.427 (>80) which is acceptable. Z plot on ProSA server gave a Z-score of -4.05 for the 3D structure validation [Fig. 5B] within the acceptable range for known structures of this size. QMEAN4 score was 1.25 which is considered optimal [Fig. 5E]. The overall model is considered suitable for further analysis. The antigenicity assessed by ANTIGENpro server was 0.6918 (>0.5) which is acceptable. Similarly, the Allergencity prediction by both AllerTop 2.0 and AllergenFP 1.0 showed the multi-epitope vaccine construct to be a non-allergen. As per AllergenFP 1.0 server and AllerTop 2.0. ToxinPred predicted the vaccine candidate sequence to be non-toxic.
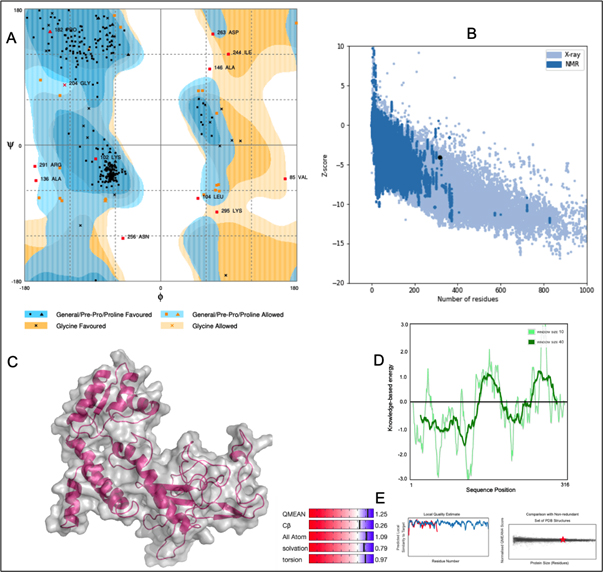
Fig. 5. (A) RAMPAGE derived Ramachandran plot for Multi-epitope vaccine construct (B) Z-plot showing the 3D model score to be within known structures derived through X-ray and NMR (C) 3D model of the chimeric vaccine construct (D) Energy per residue (E) QMEAN4 analysis of the vaccine construct.

Fig. 6. Secondary structure prediction by PSPIRED server
Physiochemical properties and secondary structure
The whole construct has a molecular weight of 33481.64 Daltons with a Theoretical isoelectric point of 8.33. The instability index (II) was calculated to be 27.07 (<40) which indicates that the protein is stable post expression [Table 3]. The extinction coefficient is computed to be around 27055 M-1 cm-1 at 280 nm. The protein has an estimated half-life of 30 hours (mammalian reticulocytes, in-vitro), >20 hours (yeast, in-vivo) and >10 hours (Escherichia coli, in-vivo). Upon over-expression, the protein is soluble with a probability of 96.45%. The Grand average of hydropathicity (GRAVY) is 0.051 which indicates that the protein is slightly on the hydrophilic side. The diameter of the protein is 4.69979 nm with an aliphatic index of 94.53. Secondary structure of the protein comprises of 35% helix, 19% beta-sheet and 30% coils. The solvent accessibility studies reveal that the 50% of the protein is exposed while 19% is medium exposed and 30% is buried. A total of 43 (13%) positions were found to be disordered. The PSPIRED secondary structure map is shown in [Fig. 6].
Table (3):
Analysis of physiochemical, antigenic, allergenic and toxic properties.
No |
Parameter |
Assessment |
|---|---|---|
1. |
Formula |
C1515H2417N401O440S6 |
2. |
Total number of atoms |
4779 |
3. |
Molecular weight |
33481.64 Daltons |
4. |
Theoretical pI |
8.33 |
5. |
Extinction coefficient measured in water |
27055 M-1 cm-1 |
6. |
Instability Index |
27.07 |
7. |
Total number of negatively charged residues (Asp + Glu): |
35 |
8. |
Total number of positively charged residues (Arg + Lys) |
37 |
9. |
Aliphatic index |
94.53 |
10. |
Grand average of hydropathicity (GRAVY) |
0.051 |
11. |
Solubility upon Overexpression |
SOLUBLE with probability 0.964524 |
12. |
Antigenicity |
0.691815 |
13. |
Allergensity |
Negative |
14. |
Toxicity |
Negative |
Molecular docking interaction between Multi-epitope vaccine construct and Toll like receptor (TLR-4)
The vaccine construct interacted with TLR-4 with a binding affinity of -8.9 Kcal/mol with a dissociation constant (KD) of 3.2×10-7 at 25°C as per PRODIGY prediction [Fig. 7A]. The free energy of the complex calculated by MMGBSA was -73.28 Kcal/mol showing stability of the complex. There were nine hydrogen bonding interactions observed [Table]. The residues Leu 190, Tyr 191, Lys 242, Ile 244, Ala 245, Leu 246 and Ser 249 of the multi-epitope vaccine construct were identified as interaction hot spots. Similarly, the residues Leu 328, Val 343, Val 348, Phe 349, Leu 350, Val 351, Phe 355, Tyr 376 of the TLR-4 were also interacting hot regions. Interestingly, the demonstrated hydrogen bonding between Tyr 191 of the vaccine construct and Tyr 376 of TLR-4 was also part of the hot region binding further validating the docking simulation.
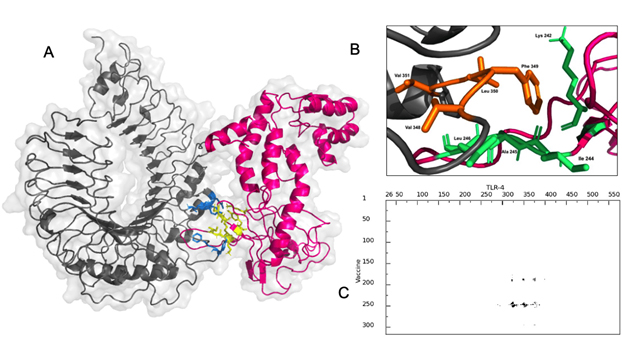
Fig. 7. (A) Interaction between Vaccine construct (pink) with binding region (blue) and TLR-4(grey) with binding region (yellow) (B) Hot regions of vaccine (green) and TLR-4 (orange) (C) interaction map between vaccine and TLR-4 by residue.
Table (4):
Constituents of Multi-epitope vaccine construct.
| No | Epitope | Position | Specificity |
|---|---|---|---|
| 1. | AMDEGYFAY | 223-231 | T-Cytotoxic cells |
| 2. | AENPVFTVF | 532-540 | |
| 3. | VGFLVRTEF | 367-375 | |
| 4. | PEICWEGVY | 500-508 | |
| 5. | QGDTLYFPAVGFLVR | 357-373 | T-Helper cells |
| 6. | PANIGLLGSKISQST | 130-145 | |
| 7. | DSKILSAFNTVIALL | 53-67 | |
| 8. | IALLGSIVIIVMNI | 64-77 | B-Cell |
| 9. | RSTDNQAV | 75-82 | |
| 10. | SCSRGVS | 239-245 | |
| 11. | LVVNWRNNTVISRPG | 485-489 | Interleukin-10 |
| 12. | PLVVNWRNNTVISRP | 484-488 |
Molecular Dynamics simulation
Normal model analysis of the complex showed mobility [Fig. 8A]. The fluctuations across the deformability plot [Fig. 9E] proves the existence of coiled structures which provides flexibility across the complex. The Eigenvalue of 3.404875×10-5 is relatively high suggesting stable complex formation [Fig. 9A]. Additionally, there is a presence of correlational elastic network along the complex as shown in [Fig. 9B] wherein the grey dots are interactions as per IMODS server. Interestingly, this elastic network is visible as atomic springs within vaccine-TLR-4 complex according to DynOmics 1.0 server further validating the increased flexible interactions [Fig. 8B]. There is an atomic mobility within the complex but the region of interaction remains stable with low mobility [Fig. 8A].
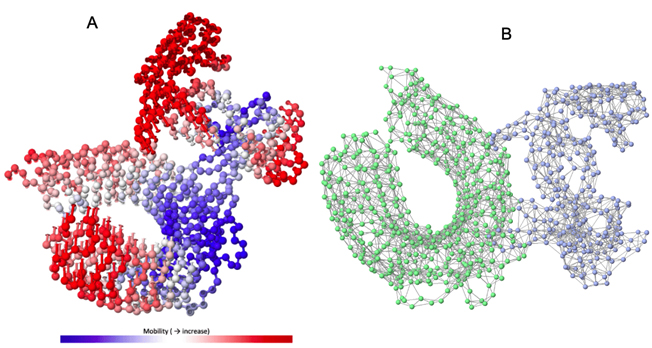
Fig. 8. (A) The mobility of Vaccine construct-TLR-4 complex (B) Elastic spring network across the atoms of vaccine-TLR-4 complex.
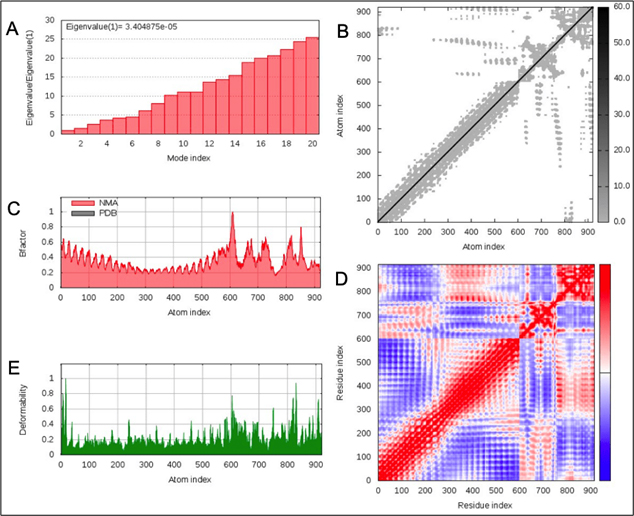
Fig. 9. (A) Eigenvalue of the vaccine-TLR-4 complex (B) Elasticity network mapping (C) Predicted experimental Bi-factor (D) Co-variance map (E) Main chain deformability.
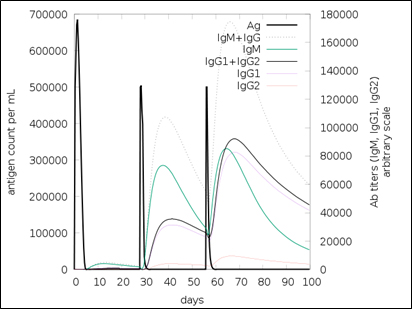
Fig. 10. Simulated antibody response upon vaccination
In silico Immune-simulation upon vaccination
The simulated immune response upon vaccine injection was similar to actual immune system [Fig. 10]. The vaccine candidate elicited IgM antibody response upon primary administration. Subsequent booster dose injections at day 30 and day 60 showed increased levels of IgG1 and IgG2 compared to IgM. Overall, the IgM and IgG antibody levels exceeded with the progressive reduction of antigen concentration after day 60 [Fig. 10]. This implies the activation of immune memory and enhanced antigen clearance upon repeated exposure. Additionally, B lymphocyte population continued to rise with the administration of the vaccine and its booster doses at day 30 and day 60 [Fig. 11A]. Of this, majority of B cell populations were found to be in active phase [Fig. 11B]. Similarly, T-Helper populations increased with the administration of the chimeric vaccine with majority of them being in the active phase [Fig. 11D]. Interestingly, T-Helper (memory) cell levels remained stable in spite of a reduction in non-memory type T-Helper cells [Fig. 11D]. T-cytotoxic cell populations gradually rose with a bulk of them being in the active phase [Fig. 12A]. The dendritic cells, Macrophages and epithelial cell populations remain constant throughout but the amount of activation slightly increased [Fig. 12C,12D and 12E]. Importantly, the enhanced expression of cytokines such as interleukins and interferons are a key to induce anti-viral response [Fig.12F]. Particularly, IFN-γ and interleukin-2 levels showed increased production while slight interleukin-10 concentrations were also observed [Fig. 12F]. Overall, immune simulation indicated a distinct secondary immune response visible post vaccination demonstrated its utility.
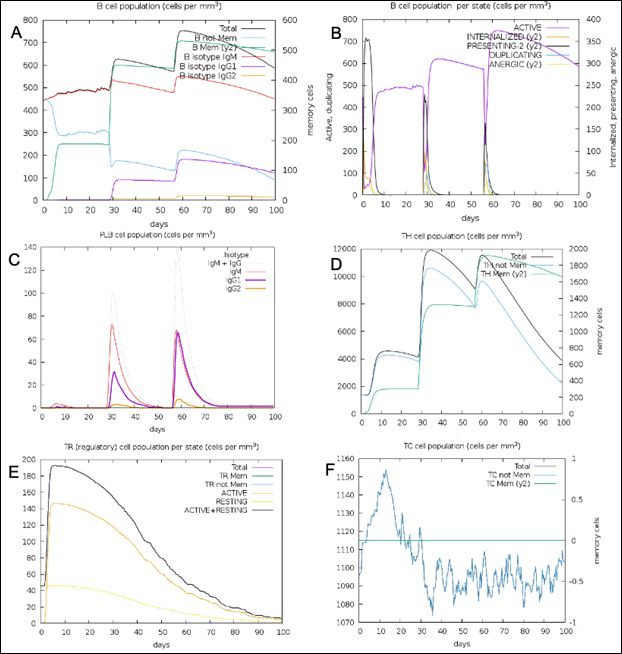
Fig. 11. (A) B-lymphocytes total count (B) B-lymphocyte population per state (C) Plasma B-lymphocytes total count (D) T-Helper cell total count (E) T-Regulatory cells per state (F) T-cell total population
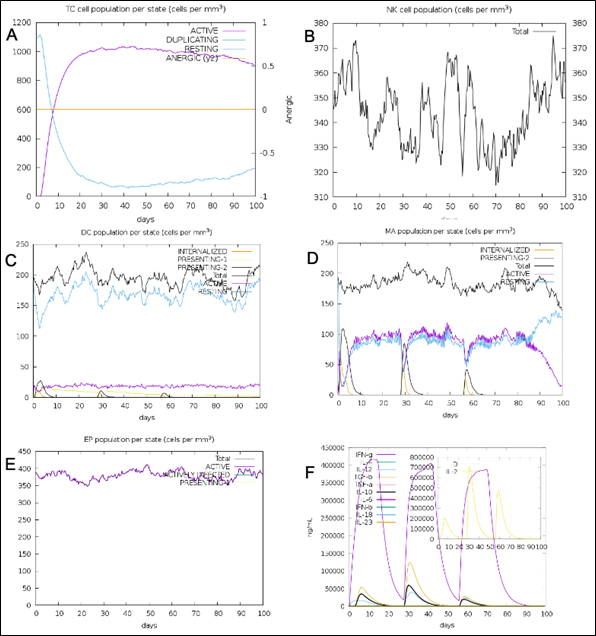
Fig. 12. (A) T-cell population per state (B) NK cell total count (C) Dendritic cell population per state (D) Macrophage population per state (E) Epithelial cell population per state (F) Cytokines concentration including interleukins and interferons.
In-silico codon optimization and cloning
The optimized gene sequence for vaccine construct was obtained using Java codon optimization tool (JCat). The adapted sequence showed 50.73% GC content which is within acceptable range (30-50%). The Codon Adaptation Index (CAI) value for the sequence was derived to be 0.95 out of 1.0 (0.8-1.0), it implies the improved sequence will show high expression levels of the chimeric vaccine protein within Escherichia coli K12 strain host. The gene sequence of 960bp was inserted in between the restriction sites HindIII and NdeI and cloned in PUC19 vector using SnapGene tool [Fig. 13].
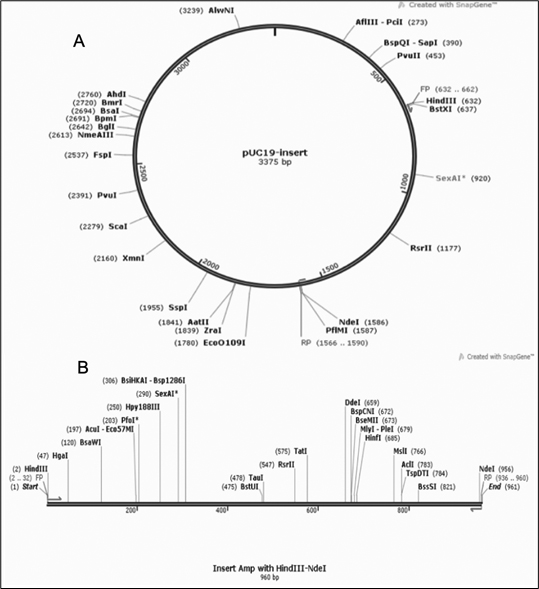
Fig. 13. (A) gene insert in PUC19 vector (B) Insert map with gene of interest in between HindIII and NdeI restriction sites.
Nipah virus is an infectious virus with high zoonotic potential and mortality60. Patients suffer with permanent neurological conditions such as encephalopathy, facial paralysis, cerebral atrophy and cervical dystonia post recovery60,61. The mortality rate in recent outbreaks varied between 32-92%60. Many broad-spectrum therapies have been developed and tested in the animal models. The drug ‘Favipiravir’ shows good recovery rate while other G-protein specific antibodies are also in the pipeline60. However, these therapies cannot undo the existing irreversible neurological damage caused by NiV. In that case, a viral specific vaccine offers a promising solution to stimulate the immune system prior to the actual infection. Many approaches were used to develop a suitable vaccine. A subunit epitope-based vaccine development remains a good alternative as the risk associated with virulence and incomplete attenuation is completely eliminated. Epitope based vaccines for Nipah virus were first derived by Sakib et al. from NiV F and G-glycoproteins62. The core focus of this paper was not just limited to deriving antigenic epitopes but also validating its role in eliciting the immune response. We isolated five T-Cytotoxic cell specific epitopes from G-protein after immunogenic assessment [Table1]. The epitopes were validated for their interaction with HLA-B*4402 which is known to cause antiviral response and facilitates interleukin-10 secretion21. Homology modelling technique was employed to design the 3D models for the epitopes as well as the MHC-1 receptor. The refined structure showed 96.2% of the residues to be in the favored region while 2.6% in the allowed and 0.7% to be in outlier region. Molecular docking simulation between HLA-B*4402 and AENPVFTVF showed stable interaction with binding affinity of -6.9 Kcal/mol and KD of 4.7×10-7. This interaction was confirmed in immune-simulation wherein the epitope was predicted to interact with HLA-B*4402 [Supplementary report 1]. T-Helper cell specific epitopes were obtained using NetMHCII 2.3 server wherein out of 534 epitopes 13 epitopes were strong binders. Out of these, three epitopes QGDTLYFPAVGFLVRT, PANIGLLGSKISQST and DSKILSAFNTVIALL were used in the vaccine construct. The immune-simulation clearly demonstrated the exceeding presence of T-Helper (memory) after day 60 while the non-memory type T-Helper cell levels gradually declined [Fig. 11F]. This proves the function of T-Helper cell specific epitopes used. Similarly, a total of 28 B-cell specific epitopes were identified from which IALLGSIVIIVMNI peptide was chosen from SVMTriP server. The B-cell epitopes RSTDNQAV and SCSRGVS were selected using BepiPred Linear epitope prediction 2.0 method on IEDB. IFN-γ eliciting epitopes were 88 from which LVVNWRNNTVISRPG and PLVVNWRNNTVISRP were chosen. The immune-simulation showed an incremental increase in B-cell response upon injection at day 0, 30 and 60. Majority of these B-cells were in the active phase. This implies the activity B-cell specific epitopes to induce precise response. A similar spike in IFN-γ concentration was seen due to the action of epitopes used in the vaccine design [Fig. 12F]. TLR-3 and TLR-4 are well known to prompt an anti-viral response. A TLR-4 targeted adjuvant was thus included in the chimeric vaccine to stimulate the humoral and cell mediated immune system 63. A multiepitope vaccine candidate was designed employing homology modelling method. In-silico structural studies on the epitope vaccine revealed adequate conformance with 281 (89.5%) in favored, 21 (6.7%) in the allowed and 12 (3.8%) in the outlier regions respectively. The Z-plot showed the presence of the structural features of the vaccine candidate similar to the in vitro structures analyzed by X-ray and Nuclear magnetic resonance (NMR) based techniques [Fig. 5B]. A docking simulation of the chimeric vaccine candidate with TLR-4 was performed to test the binding potential. The binding affinity was predicted to be -8.9 Kcal/mol while the KD was 3.2×10-7 at 25°C as per PRODIGY prediction. The binding free energy of the complex was -73.28 kcal/mol which is highly stable. Molecular dynamics simulation revealed the presence of an elastic network across atoms in the complex [Fig. 8B]. Interestingly, there was no atomic mobility observed at the binding regions within the complex indicating a firm interaction. This is validated by the relatively high eigenvalue of 3.404875×10-5 observed. Immune simulation also revealed the elevated presence of dendritic cell and macrophage populations to be around 200 cells/mm3 which remained consistent [Fig. 12C,12D]. While the natural killer cell populations fluctuated with the administration of booster doses [Fig. 12B]. To confirm the potency of the vaccine candidate, immunoreactivity needs be observed in serological analysis. This requires the presence of protein sample to perform various in-vitro and in-vivo assays. To boost the expression of the protein in a suitable bacterial system such as Escherichia coli K12 strain, codon optimization was performed which showed a CAI score of 0.95 (0.8-1.0). This implies a stable expression of chimeric protein within the bacterial host. Similarly, a poly-histidine tag (6xHis) tag was added to ease protein purification step using widely available Nickel- Nitrilotriacetic acid (Ni-NTA) agarose column [Fig. 4]. The current vaccine candidate showed promise in in-silico analysis. However, further investigation is mandated to confirm the efficacy in-vivo and in-vitro.
To control the spread of Nipah virus many aspects of public health must be strengthened. These include diagnostics, treatment and prevention. Prophylactic measures such as vaccines and mathematical outbreak prediction can reduce the mortality rates associated with the viral spread. The in-silico epitope based chimeric vaccine hopes to supplement this effort. The vaccine candidate successfully stimulated both humoral and cell mediated response in immune-simulation. A serological analysis including in-vitro and in-vivo are needed to continue the vaccine development process. This approach can also be applied to understand and develop a vaccine against Hendra virus.
Additional file: Additional Table S1.
ACKNOWLEDGMENTS
None.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
SR, VKB and DS conceived and executed the idea. VKB derived epitopes using immunoinformatics. SR performed docking and molecular dynamics simulation. Both SR and VKB performed immune-simulation analysis. VKB and SR compiled the data, illustrated the Figures and tables. SR, DS and VKB wrote the manuscript. DS supervised the whole work. All authors approved the final manuscript.
FUNDING
None.
ETHICS STATEMENT
Not applicable.
AVAILABILITY OF DATA
All datasets generated or analyzed during this study are included in the manuscript and/or the Supplementary Files.
- Bellini WJ, Harcourt BH, Bowden N, Rota PA. Nipah virus: an emergent paramyxovirus causing severe encephalitis in humans. J Neurovirol. 2005;11(5):481-487.
Crossref - Yob JM, Field H, Rashdi AM, et al. Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerg Infect Dis. 2001;7(3):439-441.
Crossref - Chatterjee P. Nipah virus outbreak in India. The Lancet. 2018;391(10136):2200.
Crossref - Clayton BA. Nipah virus: transmission of a zoonotic paramyxovirus. Curr Opin Virol. 2017;22:97-104.
Crossref - Ksiazek TG, Rota PA, Rollin PE. A review of Nipah and Hendra viruses with an historical aside. Virus Res. 2011;162(1-2):173-183.
Crossref - Arunkumar G, Chandni R, Mourya DT, et al. Nipah Investigators People and Health Study Group. Outbreak investigation of Nipah virus disease in Kerala, India, 2018. The Journal of Infectious Diseases. 2019;219(12):1867-1878.
Crossref - Wang LF, Harcourt BH, Yu M, et al. Molecular biology of Hendra and Nipah viruses. Microbes and Infection. 2001;3(4):279-287.
Crossref - Anderson DE, Islam A, Crameri G, et al. Isolation and full-genome characterization of Nipah viruses from bats, Bangladesh. Emerg Infect Dis. 2019;25(1):166-170.
Crossref - Hino K, Sato H, Sugai A, Kato M, Yoneda M, Kai C. Downregulation of Nipah virus N mRNA occurs through interaction between its 3′ untranslated region and hnRNP D. J Virol. 2013;87(12):6582-6588.
Crossref - Sun B, Jia L, Liang B, Chen Q, Liu D. Phylogeography, transmission, and viral proteins of Nipah virus. Virol Sin. 2018;33(5):385-393.
Crossref - Yoneda M, Guillaume V, Sato H, et al. The nonstructural proteins of Nipah virus play a key role in pathogenicity in experimentally infected animals. PLoS One. 2010;5(9):e12709.
Crossref - Vogt C, Eickmann M, Diederich S, Moll M, Maisner A. Endocytosis of the Nipah virus glycoproteins. J Virol. 2005;79(6):3865-3872.
Crossref - Diederich S, MAISNER A. Molecular characteristics of the Nipah virus glycoproteins. Ann N Y Acad Sci. 2007;1102(1):39-50.
Crossref - Ang BS, Lim TC, Wang L. Nipah virus infection. J Clin Microbiol. 2018;56(6):e01875-17.
Crossref - Ramphul K, Mejias SG, Agumadu VC, Sombans S, Sonaye R, Lohana P. The killer virus called Nipah: a review. Cureus. 2018;10(8):e3168.
Crossref - He Y, Rappuoli R, De Groot AS, Chen RT. Emerging vaccine informatics. J Biomed Biotechnol. 2010; 2010:218590.
Crossref - Esser MT, Marchese RD, Kierstead LS, et al. Memory T cells and vaccines. Vaccine. 2003;21(5-6):419-430.
Crossref - Doytchinova IA, Flower DR. VaxiJen: a server for prediction of protective antigens, tumour antigens and subunit vaccines. BMC Bioinformatics. 2007:8;4.
Crossref - Larsen MV, Lundegaard C, Lamberth K, Buus S, Lund O, Nielsen M. Large-scale validation of methods for cytotoxic T-lymphocyte epitope prediction. BMC Bioinformatics. 2007;8;424.
Crossref - Nielsen M, Lundegaard C, Worning P, et al. Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci. 2003;12(5):1007-1017.
Crossref - Khanna R, Burrows SR, Neisig A, Neefjes J, Moss DJ, Silins SL. Hierarchy of Epstein-Barr virus-specific cytotoxic T-cell responses in individuals carrying different subtypes of an HLA allele: implications for epitope-based antiviral vaccines. J Virol. 1997;71(10):7429-7235.
Crossref - Lamiable A, Thevenet P, Rey J, Vavrusa M, Derreumaux P, Tuffery P. PEP-FOLD3: faster de novo structure prediction for linear peptides in solution and in complex. Nucleic Acids Res. 2016;44(W1): W449-454.
Crossref - Roy A, Kucukural A, Zhang Y. I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc. 2010;5(4):725-738.
Crossref - Xu D, Zhang Y. Improving the physical realism and structural accuracy of protein models by a two-step atomic-level energy minimization. Biophys J. 2011;101(10):2525-2534.
Crossref - Lovell SC, Davis IW, Arendall III WB, et al. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins: Structure, Function, and Bioinformatics. 2003;50(3):437-450.
Crossref - Colovos C, Yeates TO. Verification of protein structures: patterns of non-bonded atomic interactions. Protein Scie. 1993;2(9):1511-1519.
Crossref - Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26(2):283-291.
Crossref - Jimenez J, Doerr S, Martinez-Rosell G, Rose AS, De Fabritiis G. DeepSite: protein-binding site predictor using 3D-convolutional neural networks. Bioinformatics. 2017;33(19):3036-3042.
Crossref - Pettersen EF, Goddard TD, Huang CC, et al. UCSF Chimera-a visualization system for exploratory research and analysis. J Comput Chem. 2004;25(13):1605-1612.
Crossref - Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2):455-461.
Crossref - Xue LC, Rodrigues JP, Kastritis PL, Bonvin AM, Vangone A. PRODIGY: a web server for predicting the binding affinity of protein-protein complexes. Bioinformatics. 2016;32(23):3676-3678.
Crossref - Weng G, Wang E, Wang Z, et al. HawkDock: a web server to predict and analyze the protein-protein complex based on computational docking and MM/GBSA. Nucleic Acids Res. 2019;47(W1):W322-W330.
Crossref - Nielsen M, Lund O. NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics. 2009;10(1):296.
Crossref - Larsen JEP, Lund O, Nielsen M. Improved method for predicting linear B-cell epitopes. Immunome Res. 2006;2:2.
Crossref - Yao B, Zhang L, Liang S, Zhang C. SVMTriP: a method to predict antigenic epitopes using support vector machine to integrate tri-peptide similarity and propensity. PLoS one. 2012;7(9):e45152.
Crossref - Dhanda SK, Vir P, Raghava GP. Designing of interferon-gamma inducing MHC class-II binders. Biology Direct. 2013;8:30.
Crossref - Lee SJ, Shin SJ, Lee MH, et al. A potential protein adjuvant derived from Mycobacterium tuberculosis Rv0652 enhances dendritic cells-based tumor immunotherapy. PLoS one. 2014;9(8):e104351.
Crossref - Shey RA, Ghogomu SM, Esoh KK, et al. In-silico design of a multi-epitope vaccine candidate against onchocerciasis and related filarial diseases. Sci Rep. 2019;9(1):4409.
Crossref - Hengen PN. Purification of His-Tag fusion proteins from Escherichia coli. Trends in Biochem Sci. 1995;20(7):285-286.
Crossref - Kelley LA, Mezulis S, Yates CM, Wass MN, Sternberg MJ. The Phyre2 web portal for protein modelling, prediction and analysis. Nat Protoc. 2015;10(6):845-858.
Crossref - Bhattacharya D, Nowotny J, Cao R, Cheng J. 3Drefine: an interactive web server for efficient protein structure refinement. Nucleic Acids Research. 2016;44(W1): W406-W409.
Crossref - Heo L, Park H, Seok C. GalaxyRefine: protein structure refinement driven by side-chain repacking. Nucleic Acids Research. 2013;41(W1):W384-W388.
Crossref - Benkert P, Biasini M, Schwede T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics. 2011;27(3):343-350.
Crossref - Wiederstein M, Sippl MJ. ProSA-web: interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Research. 2007;35(suppl_2): W407-W410.
Crossref - Magnan CN, Zeller M, Kayala MA, et al. High-throughput prediction of protein antigenicity using protein microarray data. Bioinformatics. 2010;26(23):2936-2943.
Crossref - Dimitrov I, Naneva L, Doytchinova I, Bangov I. AllergenFP: allergenicity prediction by descriptor fingerprints. Bioinformatics. 2014;30(6):846-851.
Crossref - Dimitrov I, Bangov I, Flower DR, Doytchinova I. AllerTOP v. 2-a server for in silico prediction of allergens. J Mol Model. 2014;20(6):2278.
Crossref - Gupta S, Kapoor P, Chaudhary K, Gautam A, Kumar R, Raghava GP. Open Source Drug Discovery Consortium. In silico approach for predicting toxicity of peptides and proteins. PloS one. 2013;8(9): e73957.
Crossref - Jones DT. Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol. 1999;292(2):195-202.
Crossref - Wang S, Li W, Liu S, Xu J. RaptorX-Property: a web server for protein structure property prediction. Nucleic Acids Res. 2016;44(W1):W430-W435.
Crossref - Cheng J, Baldi P. Three-stage prediction of protein β-sheets by neural networks, alignments and graph algorithms. Bioinformatics. 2005;21(suppl_1):75-84.
Crossref - Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, Vajda S. The ClusPro web server for protein-protein docking. Nat Protoc. 2017;12(2):255-278.
Crossref - Vangone A, Spinelli R, Scarano V, Cavallo L, Oliva R. COCOMAPS: a web application to analyze and visualize contacts at the interface of biomolecular complexes. Bioinformatics. 2011;27(20):2915-2916.
Crossref - Cukuroglu E, Gursoy A, Keskin O. HotRegion: a database of predicted hot spot clusters. Nucleic Acids Res. 2012;40(D1):D829-D833.
Crossref - Lopez-Blanco JR, Aliaga JI, Quintana-Orti ES, Chacon P. iMODS: internal coordinates normal mode analysis server. Nucleic Acids Res. 2014;42(W1):W271-W276.
Crossref - Yang LW, Liu X, Jursa CJ, et al. i GNM: a database of protein functional motions based on Gaussian Network Model. Bioinformatics. 2005;21(13):2978-2987.
Crossref - Rapin N, Lund O, Bernaschi M, Castiglione F. Computational immunology meets bioinformatics: the use of prediction tools for molecular binding in the simulation of the immune system. PLoS One. 2010;5(4):e9862.
Crossref - Grote A, Hiller K, Scheer M, Munch R, Nortemann B, Hempel DC, Jahn D. JCat: a novel tool to adapt codon usage of a target gene to its potential expression host. Nucleic Acids Res. 2005;33(suppl_2): W526-W531.
Crossref - Biotech G. SnapGene Viewer. Glick B, editor. 2020;3(3). (www.snapgene.com) [Online, Accessed on 12 June 2020].
- Gasteiger E, Hoogland C, Gattiker A, Wilkins MR, Appel RD, Bairoch A. Protein identification and analysis tools on the ExPASy server. In the proteomics protocols handbook. 2005:571-607. Humana Press.
Crossref - Magnan CN, Randall A, Baldi P. SOLpro: accurate sequence-based prediction of protein solubility. Bioinformatics. 2009;25(17):2200-2207.
Crossref - Singh RK, Dhama K, Chakraborty S, et al. Nipah virus: epidemiology, pathology, immunobiology and advances in diagnosis, vaccine designing and control strategies-a comprehensive review. Vet Quarterly. 2019;39(1):26-55.
Crossref - Sejvar JJ, Hossain J, Saha SK, et al. Long-term neurological and functional outcome in Nipah virus infection. Ann Neurol: Official Journal of the American Neurological Association and the Child Neurology Society. 2007;62(3):235-242.
Crossref - Sakib MS, Islam M, Hasan AK, Nabi AH. Prediction of epitope-based peptides for the utility of vaccine development from fusion and glycoprotein of nipah virus using in silico approach. Adv Bioinformatics. 2014;2014:403492.
Crossref - Vaidya SA, Cheng G. Toll-like receptors and innate antiviral responses. Curr opin Immunol. 2003;15(4):402-407.
Crossref - El-Manzalawy Y, Dobbs D, Honavar V. Predicting flexible length linear B-cell epitopes. Computational Systems Bioinformatics. 2008;7:121-132.
Crossref - Hubbard RE, Haider MK. Hydrogen bonds in proteins: role and strength. Encyclopedia of Life Sciences. 2010;1-7.
Crossref
© The Author(s) 2021. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.