


ISSN: 0973-7510
E-ISSN: 2581-690X
The purpose of the research was to obtain different variants of chickpea beverages and subject them to probiotic fermentation carried out by Lactobacillus plantarum 299v in order to compare properties of received products. The study focused on the influence of the fermentation process on changes in protein profiles, antioxidative properties, pH values, and reducing power (RP). The analyzed variants of chickpea beverages exhibited diversity in terms of RP. Fermented products were characterized by significantly higher values of RP (A700) ranging from 0.1 to 0.29 than unfermented ones (value in range from 0.04 to 0.09). Whereas, pH values of fermented products were lower (from 4.46 to 5.64) to than unfermented beverages (6.68 to 7.57). The results indicate also the presence of b-conglycinin and glycinin (which are considered as one of food allergens) in analyzed chickpea-based beverages, however the probiotic fermentation contributed to strong hydrolysis of those components. Among tested variants of the chickpea products, the beverages with vanillin sugar and coconut flakes (unfermented as well as fermented) received the highest notes of general consumer acceptance in preliminary sensory assessment. The fermentation of the chickpea beverages with the probiotic strain significantly increased the reducing power of the products, revealed antioxidative-enhancing properties and contributed to hydrolysis of b-conglycinin (7S) and glycinin (11S) fractions.
Perna perna, bacterial populations, phylogeny, sequencing, 16S rRNA gene.
Bivalve molluscs are economically important marine shellfish which are abundantly distributed in estuarine and marine waters whose temperature remains below 30°C. They are usually found on rocky shores in temperate regions1, thus they are prone to contamination by toxin-producing dinoflagellates and faecal pathogens2,3.
Perna perna L. is an indigenous mussel which proliferates on the rocky reefs along the eastern coast of South Africa. It is also indigenous to the waters of Namibia, Angola, Cape Verde, Mauritania, Mozambique, Madagascar, Europe, South and North America up to the Caribbean4,5.
According to Potasman et al.3, this organism serves as a significant source of food in different parts of the world. The consumption of Perna perna is associated with health benefits including neurologic development during gestation and infancy6, reduced the risk of heart diseases and lowering the amount of cholesterol in the blood7. Other benefits include the treatment of rheumatoid arthritis, improved circulation and reduction of the risk of thrombosis. It also provides the body with many essential nutrients including iodine, selenium, zinc and potassium8. Despite these benefits, there is a potential risk of bacterial infection attributed to the consumption of mussels. Since they are filter feeders, they concentrate microorganisms in their guts and are therefore regarded as potential reservoirs of diverse microbial pathogens9.
To the best of our knowledge, phylogenetic profiling of bacterial populations in Perna perna from Algoa Bay, Port Elizabeth, Eastern Cape Province of South Africa has not been investigated despite the mass consumption of the organism. In light of this, the study aimed at identifying and profiling different bacterial populations in P. perna collected from Algoa Bay using molecular-based techniques. This is to create awareness to the consumers about the bacterial contamination of the mussels and to educate them to avoid eating raw seafood.
Description of the study area
Algoa Bay is located in Port Elizabeth, Eastern Cape Province of South Africa (33°49′ 36″ S, 25°47′ 42″ E). It is a wide inlet along the South African east coast and 425 miles east of the Cape of Good Hope. The Bay is about 436 m deep, bounded in the west by Cape Recife and to the east by Cape Padrone.
Sampling and dissection of Perna perna
Fifty Perna perna aseptically collected from the rocky shores of Algoa Bay were transported in ice packs to the Microbiology Laboratory, University of Fort Hare for dissection and examination. They were surface sterilized with 70% ethanol, rinsed three times with sterile distilled water after which the shells were then opened using a sterile scalpel blade. The shell liquid of the mussels was discarded and tissues were removed for further analysis.
DNA Extraction
Each of the Perna perna samples was homogenized in a sterile 40 ml tissue blender. About 0.5 ml of homogenate was used for the bacteria DNA extraction using Thermo Scientific GeneJET Genomic DNA Purification kit (EU, Lithuania). The DNAs were quantified using NanoDrop 2000 (Thermo Scientific, USA) and used as templates in the PCR analysis.
PCR confirmation of the targeted base pair
The bacterial 16S rRNA gene fragments were PCR-amplified using universal conserved region primers (Inqaba Biotech, SA). The nucleotide sequences of the primers are as follows: GM5F (5′-CCTACGGGAGGCAGCAG-3′) and 907R (5′-CCGTCAATTCCTTTGAGTTT-3′) with a 40 bp GC clamp CGCCCGCCGCGCCCCGCGCCCGTCCC-GCCGCCCCCGCCCG attached to the 5′ end of the reverse primer10. These primers are highly genus-specific with expected amplicon size of 586 bp. PCR amplification was carried out in 25 ml total reaction volumes consisting of 12.5 ml Master Mix (Thermo Scientific, (EU) Lithuania), 1 ml each of oligonucleotide primer (Inqaba Biotech, SA), 5 ml of template DNA and 5.5 ml of nuclease-free water. The amplification was performed on a Bio-Rad Mycycler USA thermo-cycler on the following cycling conditions: initial denaturation at 95°C for 1 min, followed by 40 cycles of denaturation at 94°C for 30 sec, annealing at 64°C for 45 sec, extension at 72°C for 1 min, and a finally extension step at 72°C for 10 mins. In order to confirm the size of the product, 2 µl of the amplicons were analyzed by gel electrophoresis in 1% agarose (Merck, SA) stained with 3 µl ethidium bromide (Sigma-Aldrich, USA). A 100 bp DNA ladder (Thermo Scientific, (EU) Lithuania) was included for band size estimation purposes. All gels were run in 0.5X TBE buffer at 95 V for 1 h and visualized by UV trans-illumination (Alliance 4.7, France).
Purification of amplicons and sequencing
PCR products were purified following the protocols of the ZymocleanTM Gel DNA Recovery kit (Zymo Research Corporation, Irvine, US). The purified DNA was eluted from spin columns with 30 µl of nuclease-free water and DNA concentrations were quantified using NanoDrop 2000 (Thermo Scientific, USA). Ten microliters of the eluate were used as the DNA template in the PCR, using the primers and conditions described above. The DNA samples were sent to Inqaba Biotech (South Africa) for sequencing analysis. The thermo-cycling reaction consisted of DNA template, 1 ml of 10 mM GM5F (52 -CCTACGGGAGGCAGCAG-3′ ) (Inqaba Biotech, SA) primer stock solution, 4ml Big Dye, 2 ml 5 x dilution buffer and distilled water (to the volume of 20 ml). The reaction samples were directly sequenced with an ABI 3500 XL genetic analysers using an ABI V3.1 Big Dye kit (Zymo Research Corporation, Irvine, US).
Phylogenetic analysis
The closest known relatives of the partial 16S rRNA sequences for the obtained microbial sequences was determined through empirical searches in the GenBank with National Center for Biotechnology Information (NCBI) database using the Basic Local Alignment Search Tool (BLAST) program. The sequences were aligned using Clustal Omega version 1.2.1 and classified into groupings based on the sequence similarities of their ribosomal database project. The 16S rRNA sequences obtained were inferred using the Neighbour-Joining method11) The evolutionary analyses and distances were computed using the Maximum Composite Likelihood method on MEGA 6 software respectively12-14. The analysis involving 36 nucleotide sequences are in the units of the number of base substitutions per site. Codon positions included were 1st, 2nd, 3rd and noncoding. All positions containing gaps and missing data were eliminated and a total of 1309 positions were included in the final dataset.
PCR amplification and identification of bacterial populations in Perna perna
The PCR amplification result revealed the expected amplicon size of 586 bp in almost all the samples (Fig. 1). A total of 28 bacterial strains were identified in samples of which 89.30% of the isolated bacteria belonged to the Phylum Actinobacteria and were all Gram-positive while the remaining 10.70% belonged to the Phylum Spirochaeta and they were Gram-negative (Table 1; Fig. 2). The comparative16S rRNA gene sequence analysis revealed that the bacterial isolates were predominantly members of the genera Saccharothrix with six species (21%) followed by Kocuria and Streptomyces with five species respectively (18%). Also, species of Spirochaeta (11%), Arthrobacter (7%), Rhodococcus (7%), Zhihengliuella (4%), Nesterenkonia (4%), Lechevalieria (4%), Citricoccus (3%) and Micrococcus (3%) were identified in the samples as shown in Fig. 2.
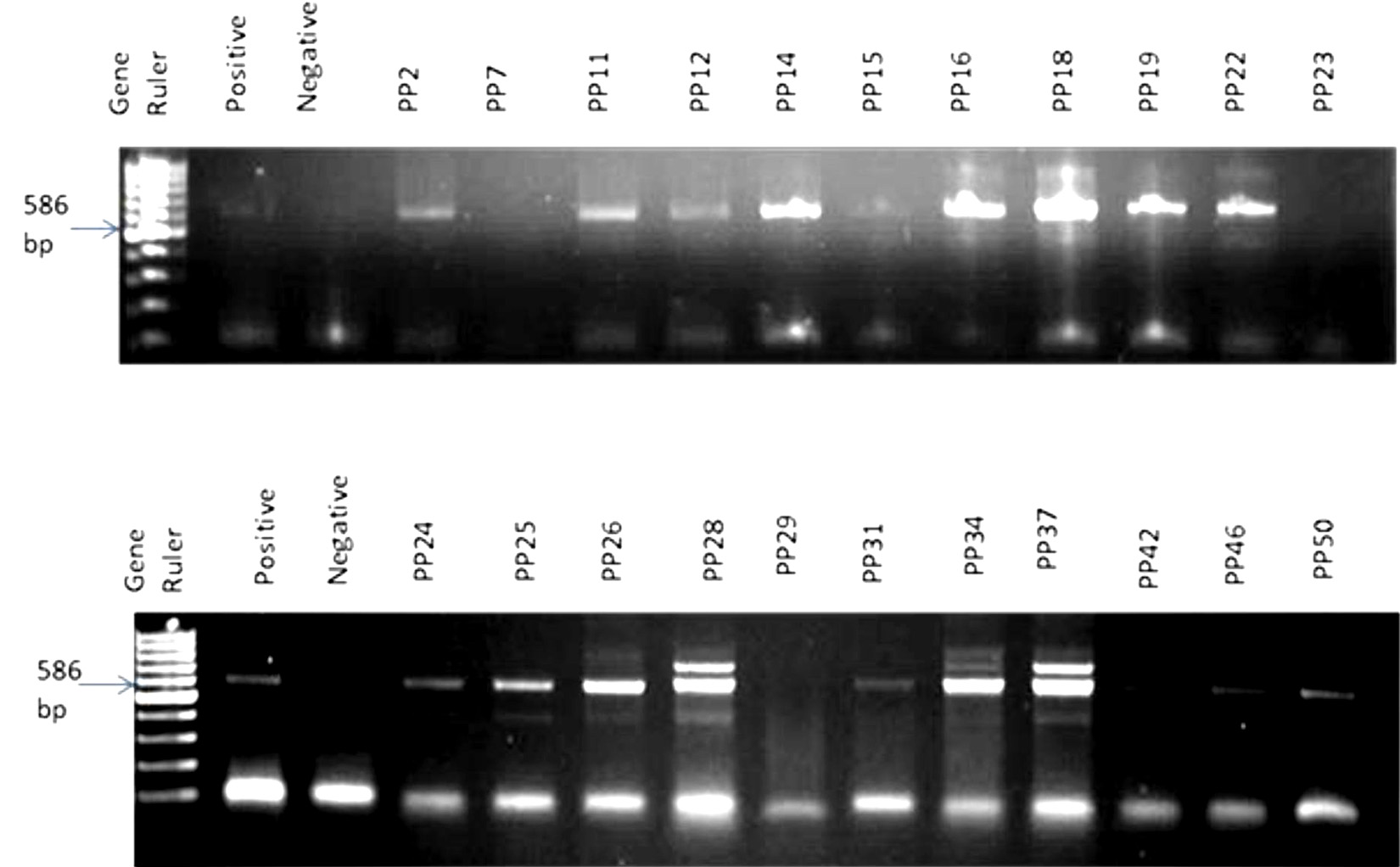
Fig. 1. A random representative gel electrophoresis of PCR confirmation of bacteria in Perna perna. Gel 1: Lanes 1: 100 bp DNA ladder, 2: positive control; 3: negative control, 4: PP2, 5:PP7, 6:PP11, 7:PP12, 8:PP14, 9:PP15, 10:PP16, 11:PP18, 12:PP19, 13:PP22, 14:PP23. Gel 2: Lanes 1: 100 bp DNA ladder, 2: positive control; 3: negative control, 4: PP24, 5:PP25, 6:PP26, 7:PP28, 8:PP29, 9:PP31, 10:PP34, 11:PP37, 12:PP42, 13:PP46, 14:PP50.
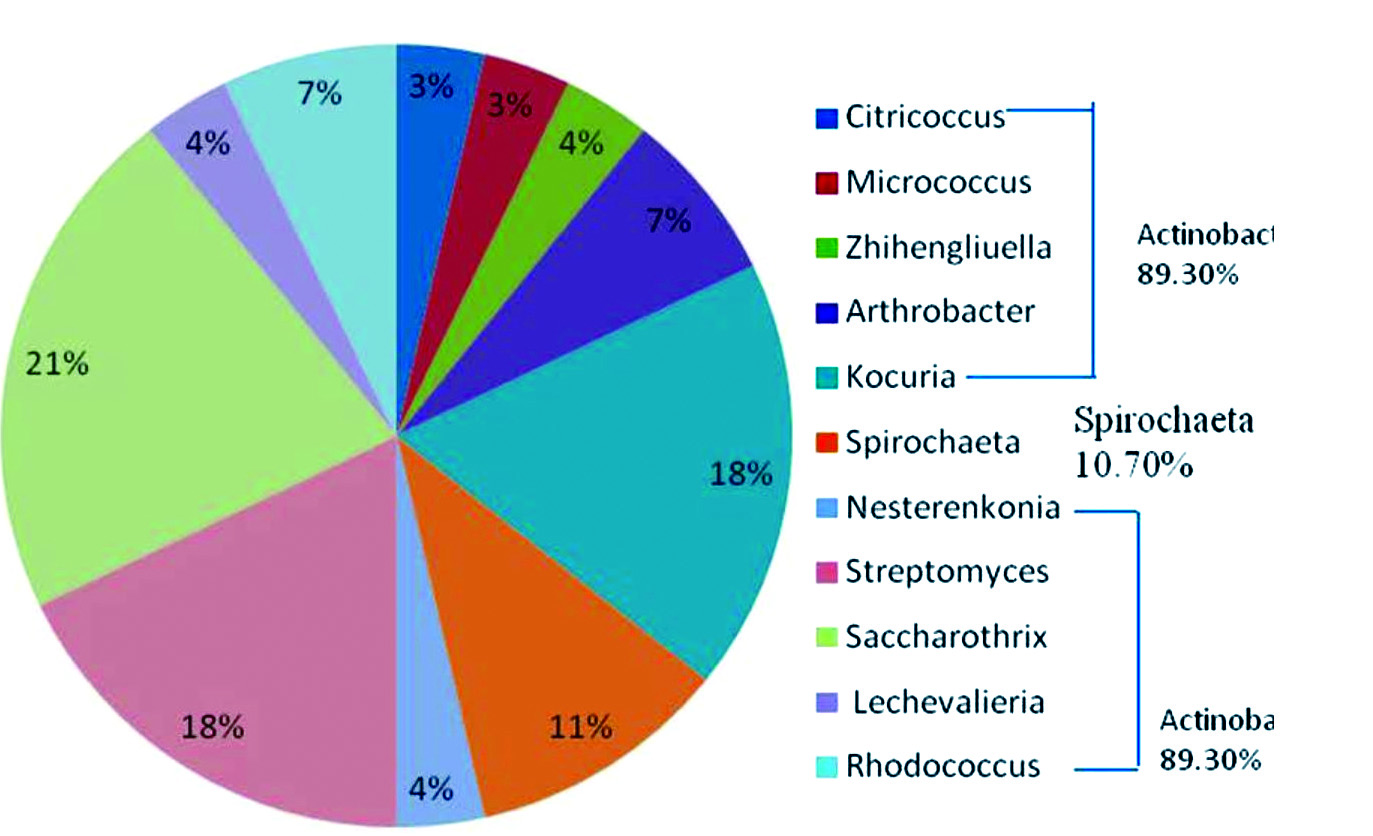
Fig. 2.Generic compositions of bacteria in Perna perna
Table (1):
Bacterial populations identified found in Perna perna
Strain |
Family |
Genus |
Species |
Gram +ve/-ve |
|---|---|---|---|---|
PNP1 |
Micrococcaceae |
Citricoccus |
nitrophenolicus |
+ve |
V3M1 |
Micrococcaceae |
Micrococcus |
terreus |
+ve |
DY66 |
Micrococcaceae |
Zhihengliuella |
aestuarii |
+ve |
CA15-8 |
Micrococcaceae |
Arthrobacter |
koreensis |
+ve |
CF-25 |
Micrococcaceae |
Arthrobacter |
luteolus |
+ve |
DSM 20447 |
Micrococcaceae |
Kocuria |
rosea |
+ve |
CAAS 251 |
Micrococcaceae |
Nesterenkonia |
flava |
+ve |
TAGA27 |
Micrococcaceae |
Kocuria |
palustris |
+ve |
104 |
Micrococcaceae |
Kocuria |
salsicia |
+ve |
ATCC 15306 |
Micrococcaceae |
Kocuria |
varians |
+ve |
KMM 3905 |
Micrococcaceae |
Kocuria |
marina |
+ve |
RCQ1071 |
Streptomycetaceae |
Steptomyces |
lunalinharesii |
+ve |
LL-C19004-NS29 |
Pseudonocardiaceae |
Saccharothrix |
espanaensis |
+ve |
NJ2035 |
Pseudonocardiaceae |
Lechevalieria |
nigeriaca |
+ve |
Hhs.015 |
Pseudonocardiaceae |
Saccharothrix |
yanglingensis |
+ve |
SA 233 |
Pseudonocardiaceae |
Saccharothrix |
algeriensis |
+ve |
DSM 43752 |
Nocardiaceae |
Rhodococcus |
marinonascens |
+ve |
NRRL 15764 |
Pseudonocardiaceae |
Saccharothrix |
espanaensis |
+ve |
NRRL B-16115 |
Pseudonocardiaceae |
Saccharothrix |
coeruleofusca |
+ve |
NRRL 11239 |
Pseudonocardiaceae |
Saccharothrix |
australiensis |
+ve |
ATCC 35653 |
Nocardiaceae |
Rhodococcus |
marinonascens |
+ve |
M1463 |
Streptomycetaceae |
Streptomyces |
samsunensis |
+ve |
NBRC 101006 |
Streptomycetaceae |
Streptomyces |
hebeiensis |
+ve |
NBRC 16446 |
Streptomycetaceae |
Streptomyces |
malaysiensis |
+ve |
YIM 001 |
Streptomycetaceae |
Streptomyces |
hebeiensis |
+ve |
R 1 |
Spirochaetaceae |
Spirochaeta |
litoralis |
-ve |
SIP1 |
Spirochaetaceae |
Spirochaeta |
cellobiosiphila |
-ve |
ASpG1 |
Spirochaetaceae |
Spirochaeta |
americana |
-ve |
Phylogenetic relationships of the identified bacteria in Perna perna
The evolutionary relationships of the bacterial populations in Perna perna is depicted in the phylogenetic tree (Fig. 3). Changes in the evolutionary trend of the bacteria and the optimal tree have the sum of branch length 18.

Fig. 3. Phylogenetic tree based on 16S rRNA gene sequences, showing bacterial populations present in Perna perna. The tree was generated using the Neighbour-Joining method. The numbers at the nodes indicated the bootstrap valves (1,000 replications). The bar 0.2 indicates the nucleotide substitution per position.
Various studies have been reported on the microbiota of bivalve molluscs due to their feeding habit. The studies had shown that a large number of bacterial pathogens are concentrated in the tissues of molluscs and as such cause disease to humans15,16.
The present study revealed that Perna perna is predominantly contaminated with members of the Phylum Actinobacteria. Actinobacteria is one of the largest phyla of bacteria. Members of this group have adopted different lifestyles such as soil inhabitants, marine dwellers, pathogens, plant and gastro-intestinal commensals17. Many species of Actinobacteria have been isolated from animal faeces indicating that these bacteria can adapt and survive in any given condition18. They could be mesophilic, psychrophilic, alkalophilic and alkalotolerant19. The prevalence of Actinobacteria in the samples could be as a result of the adaptive mechanism of the organism18.
Also, Micrococcus terreus was equally identified in the samples. Skin, soil and some marine animals are the major sources of this bacteria20. Micrococci are commensals as well serve as opportunistic pathogens in immuno-compromised individuals21. The occurrence of Micrococcus terreus in water and shellfish may be linked to land pollution20.
Members of the genus Arthrobacter were first isolated from soils in various environments22. These bacteria could be mesophilic, psychrophilic, alkalophilic and alkalotolerant19. Also, Citricoccus nitrophenolicus have been isolated from a wastewater treatment plant of a chemical factory producing methyl-parathion and other pesticides23. This bacterium may have likely been released into the ocean from a nearby wastewater treatment plants and found their way into the gut of Perna perna.
Kocuria is ubiquitous in nature and frequently found as normal skin flora in humans and other mammals; except for the facultative anaerobe Kocuria kristinae, strictly aerobe Kocuria marina and Kocuria rhizophila which can proliferate anaerobically. The present study identified seven out of 18 species of Kocuria known24,25. K. rosea and K. varians have been implicated in infective endocarditis, catheter-related bacteremia, peritonitis, necrotizing mediastinitis, arthritis, central nervous system infection, pneumonia, hepatic abscess and nosocomial bloodstream infections26-29. In general, Kocuria is a human pathogen, causing diseases mostly in immunocompromised hosts. They have also been found in traditional salt-fermented seafood, marine sediment, soil and water30-32.
Actinomycetes are unique in forming spores, production of numerous antibiotics including streptomycin, neomycin, chloram-phenicol and tetracyclines33. Over 500 species of Streptomyces have been identified, with inherent ability to produce extracellular hydrolytic enzymes, thus, they are considered as agents for bioremediation but few species are pathogenic for animals, and few might cause plant diseases34.
The genus Spirochaeta is Gram-negative, free-living, saccharolytic, non-pathogenic, obligate and facultative anaerobic helical shaped bacteria. It is indigenous to aquatic environments including muds of ponds, rivers, marshes, marine and fresh waters, and halophilic species such as alkalophiles and thermophiles35,36. The current study identified three out of about 17 species of Spirochaeta that have been reported. The detection of this bacteria could be attributable to the inflow of industrial effluents into the aquatic ecosystem, as the strain was reportedly isolated from paper-mill effluents37.
The comparative 16S rRNA gene sequence analysis affiliates the bacteria to diverse phylogenetic groups. Diversity in the phylogenetic tree might be due to changes in the bacteria nucleotide sequence caused by mutation, which could be substitution, deletion, insertion or frame shift mutation. Mutation usually leads to changes in the amino acid sequences in different bacteria38.
Also, it could be deduced from the evolutionary tree that bacteria found in Perna perna come from the same origin, but the evolutionary changes have led to taxa separation over a period of time (Fig. 3). The conserved regions of the nucleotide sequences in bacteria are important sites of evolutionary divergence38. Evolution does not alter in these regions because the regions are unique to all bacteria. Changes in any conserved region as a result of mutations could alter the regulation of conserved genes, producing species-specific patterns of gene expression and leads to evolutionary changes38. The active sites of enzymes and the binding sites of protein receptors are among the most highly conserved sequence. These sequences often harbour cis-regulatory elements which constrain evolution39.
In the study, different bacteria were having a specific nucleotide sequence which led to differences under the phylogenetic evolution. These also might lead to the different coding of the amino acid which could reflect different protein structures. In turn, the protein structures and functions might lead to differences in the phylogenetic evolution. According to40, proteins are the embodiments of the transition from the one-dimensional world of sequences to the three-dimensional world of molecules capable of diverse activities. More so, evolutionary differences occur in bacteria due to errors during translation to transcription at the molecular level. The higher levels of sequence variation allow differentiation of closely related strains41 while the ability to translate DNA to protein sequences permits phylogenetic analysis of distantly related strains and more accurate sequence alignment42.
The findings of this study indicated that molluscan shellfish examined are reservoirs of various bacterial populations. It is therefore important for the government to take adequate control measures to set specific standards for the quality of seafoods. Also, the study further advice individuals who consume seafood to avoid consumption of either raw or slightly cooked Perna perna. The microbial diversity of marine bivalves should be thoroughly investigated together with the potential microorganisms producing bioactive metabolites.
We wish to thank the National Research Foundation of South Africa for sponsoring this work. We also acknowledge the South African Institute for Aquatic Biodiversity for assistance with the collection of Perna perna. Special thanks to Dr Francesca Pori for her help with the identification and dissection of samples.
The authors declare no conflict of interest.
- Calci K R, Depaola A, Burkhardt III W. Molluscan Shellfish: Oysters, Mussels, and Clams Updated September 2013. Accessed 2014/09/08.
- Wilson I G, Moore J E. Presence of Salmonella spp. and Campylobacter spp. in shellfish. Epidemiol Infect, 1996; 116(2): 147-53.
- Potasman I, Paz A, Odeh M. Infectious outbreaks associated with bivalve shellfish consumption: a worldwide perspective. Clin Infect Dis, 2002; 35(8): 921-8.
- Monirith I, Ueno D, Takahashi S, Nakata H, Sudaryanto A, Subramanian A, Karuppiah S, Ismail A, Muchtar M, Zheng J S, Richardson B J, Prudente M, Hue N D, Tana T S, Tkalin A V, Tanabe S. Asia-Pacific mussel watch: monitoring contamination of persistent organochlorine compounds in coastal waters of Asian countries. Marine Pollution Bulletin, 2003; 46(3): 281-300.
- Global Invasive Species Database Perna perna (mollusc), 2007.
- Hibbeln JR, Davis JM, Steer C, Emmett P, Rogers I, Williams C, Golding J. Maternal seafood consumption in pregnancy and neuro-developmental outcomes in childhood (ALSPAC study): an observational cohort study. The Lancet, 2007; 369(9561): 578-585.
- Mozaffarian D, Rimm E B. Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA, 2006; 296(15): 1885-99.
- Yaktine A L, Nesheim M C, editors. Seafood choices: balancing benefits and risks. National Academies Press; 2007 Apr 9.
- Iwamoto M, Ayers T, Mahon B E, Swerdlow D L. Epidemiology of seafood-associated infections in the United States. Clin Microbiol Rev, 2010; 23(2): 399-411.
- Clarke A M, Kirby R, Rose P D. Molecular microbial ecology of lignocellulose mobilisation as a carbon source in mine drainage wastewater treatment. Water SA, 2004; 30(5): 558-661.
- Saitou N, Nei M. The neighbour-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol, 1987; 4(4): 406-25.
- Tamura K, Nei M, Kumar S. Prospects for inferring very large phylogenies by using the neighbour-joining method. Proceedings of the National Academy of Sciences, 2004; 101(30): 11030-11035.
- Tamura K, Stecher G, Peterson D, Filipski A, Kumar S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol, 2013; 30(12): 2725-2729.
- Reddy MB. Basics for the Construction of Phylogenetic Trees. Web Cen Biol, 2013; 12: WMC002563.
- Romanenko L A, Uchino M, Kalinovskaya N I, Mikhailov V V. Isolation, phylogenetic analysis and screening of marine mollusc-associated bacteria for antimicrobial, hemolytic and surface activities. Microbiol Res, 2008; 163(6): 633-644.
- Gayathri V, Ganga Baheerathi C, Revathi K, Sethi S N. Identification of Gut Microflora from Green Mussel (Perna viridis).
- Ventura M, Canchaya C, Tauch A, Chandra G, Fitzgerald G F, Chater K F, van Sinderen D. Genomics of Actinobacteria: tracing the evolutionary history of an ancient phylum. Microbiol Mol Bio Rev, 2007; 71(3): 495-548.
- Jiang Y, Han L, Chen X, Yin M, Zheng D, Wang Y, Qiu S, Huang X. Diversity and bioactivity of cultivable animal fecal actinobacteria. Advances in Microbiol, 2013; 3(01):1.
- Lee J S, Lee K C, Pyun Y R, Bae K S. Arthrobacter koreensis sp. nov., a novel alkalitolerant bacterium from soil. Intern J syst Evolutionary Microbiol, 2003; 53(5): 1277-1280.
- Kocur M, Kloos W E, Schleifer K H. The genus micrococcus. In The Prokaryotes, 2006 (pp. 961-971). Springer, New York, NY.
- Smith K J, Neafie R, Yeager J, Skelton H G. Micrococcus folliculitis in HIV-1 disease. The British J Dermatol, 1999; 141(3): 558-561.
- Holt J G, Krieg N R, Sneath P H A, Staley J T, Williams S T. Bergey’s Manual of Determinative Bacteriology. Nineth Edition. Baltimore, Maryland: Williams & Wilkins, 1994; 787 pp.
- Nielsen M B, Kjeldsen K U, Ingvorsen K. Description of Citricoccus nitrophenolicus sp. nov., a para-nitrophenol degrading actino-bacterium isolated from a wastewater treatment plant and emended description of the genus Citricoccus. Altenburger et al. 2002. Antonie van Leeuwenhoek. 2011; 99(3): 489-499.
- Stackebrandt E, Frederiksen W, Garrity GM, Grimont P A, Kהmpfer P, Maiden M C, Whitman W B. Report of the ad hoc committee for the re-evaluation of the species definition in bacteriology. Intern J syst Evolutionary Microbiol, 2002; 52(3): 1043-1047.
- Takarada H, Sekine M, Kosugi H, Matsuo Y, Fujisawa T, Omata S, Harayama S. Complete genome sequence of the soil actinomycete Kocuria rhizophila. J Bacteriol, 2008; 190(12): 4139-4146.
- Kaya K E, Kurtoנlu Y, Cesur S, Bulut C, Kinikli S, Irmak H, Demirצz A P, Karakoc E. Peritonitis due to Kocuria rosea in a continuous ambulatory peritoneal dialysis case. Mikrobiyoloji bulteni, 2009; 43 (2): 335-337.
- Lee M K, Choi S H, Ryu D W. Descending necrotizing Mediastinitis caused by Kocuria rosea: a case report. BMC Infect Dis, 2013; 13(1): 475.
- Srinivasa K H, Agrawal N, Agarwal A, Manjunath C N. Dancing vegetations: Kocuria rosea endo-carditis. BMJ case reports, 2013; 2013.
- Tsai C Y, Su S H, Cheng Y H, Chou Y L, Tsai T H, Lieu A S. Kocuria varians infection associated with brain abscess: a case report. BMC Infect Dis, 2010; 10(1): 102.
- Ben-Ami R, Navon-Venezia S, Schwartz D, Carmeli Y. Infection of a ventriculoatrial shunt with phenotypically variable Staphylococcus epidermidis masquerading as polymicrobial bacteremia due to various coagulase-negative Staphylococci and Kocuria varians. J Clin Microbiol, 2003; 41(6): 2444-2447.
- Kim S B, Nedashkovskaya O I, Mikhailov V, Han S K, Kim K O, Rhee M S, Bae K S. Kocuria marina sp. nov., a novel actinobacterium isolated from marine sediment. Intern J syst Evolutionary Microbiol, 2004; 54(5): 1617-1620.
- Savini V, Catavitello C, Masciarelli G, Astolfi D, Balbinot A, Bianco A, Febbo F, D’amario C, D’antonio D. Drug sensitivity and clinical impact of members of the genus Kocuria. J Med Microbiol, 2010; 59(12): 1395-1402.
- Kieser T, Bibb M J, Buttner M J, Chater K F, Hopwood D A. Practical Streptomyces Genetics (2nd ed.). Norwich, England: John Innes Foundation, 2000.
- Euzיby J P. “Genus Streptomyces”. List of Prokaryotic names with Standing in Nomenclature. Retrieved 2008-09-28.
- Hoover R B, Pikuta E V, Bej A K, Marsic D, Whitman W B, Tang J, Krader P. Spirochaeta americana sp. nov., a new haloalkaliphilic, obligately anaerobic spirochaete isolated from soda Mono Lake in California. Intern J syst Evolutionary Microbiol, 2003; 53(3): 815-21.
- Leschine S, Paster B J, Canale-Parola E. Free-living saccharolytic spirochetes: the genus Spirochaeta. In The prokaryotes. Springer, New York, 2006; pp. 195-210.
- Luo H Y, Miao L H, Fang C, Yang P L, Wang Y , Shi P J, Fan Y L. Nesterenkonia flava sp. nov., isolated from paper-mill effluent. Intern J syst Evolutionary Microbiol, 2008; 58 (8): 1927-1930.
- Prabhakar S, Noonan J P, Pההbo S, Rubin E M. Accelerated evolution of conserved noncoding sequences in humans. Science, 2006; 314(5800): 786-786
- Gross L. Are “ultraconserved” genetic elements really indispensable?. PLoS Biol, 2007; 5(9): e253.
- Petsko G A, Ringe D. Protein structure and function. New Science Press; 2004.
- Ochman H, Wilson A C. Evolution in bacteria: evidence for a universal substitution rate in cellular genomes. J Mol Evol, 1987; 26(1-2): 74-86.
- Gupta R S. Protein phylogenies and signature sequences: a reappraisal of evolutionary relationships among archaebacteria, eubacteria, and eukaryotes. Microbiol Mol Biol Rev, 1998; 62(4): 1435-1491.
© The Author(s) 2019. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.