


ISSN: 0973-7510
E-ISSN: 2581-690X
Johne’s disease or paratuberculosis is a chronic infectious enteric disease of ruminants caused by the intracellular pathogen. The control of the Johne’s disease is hampered by lack of specific diagnostic tests. A unique ORF 843 bp encoding 30.7 kDa hypothetical protein of Mycobacterium avium subsp. paratuberculosis (MAP) was generated using PCR. The gene was cloned in frame into E. coli expression plasmid pQE 30. The recombinant plasmid designated as pQE 30.7 was transformed into E. coli M15 and induced with IPTG revealed the high level expression of 30.7 kDa protein which was confirmed by immunoblotting. Recombinant 30.7 kDa protein was then purified by Ni-NTA agarose chromatography. The polyclonal antiserum raised against purified recombinant 30.7 protein reacted with induced E. coli whole cell lysate as well as recombinant purified proteins on western blot. The 30.7 kDa expressed in the present study will prove useful as reagent in diagnostic test.
30.7 kDa, Mycobacterium avium subsp. paratuberculosis, accession no. EF 210209.
Johne’s disease (JD) or paratuberculosis is a chronic, progressive, and incurable intestinal disease caused by Mycobacterium avium subsp. paratuberculosis (Map) in domestic and wild ruminants1. The disease causes severe economic losses to agriculture industry due to reduced milk production and premature culling (Ott et al., 1999). Infected animals with clinical signs shed the organism in feaces (Clarke et al., 1997, Stehman et al., 1996) and milk (Taylor et al., 1981), resulting in greater risk for other animals as well as human exposure (Gangadhararao et al., 2013)
The progression of Map infection to different stages mainly depends on the immune status of individual animals. Any single diagnostic test is not full proof to detect Map infection at every stage. The control of JD was severely hampered due to lack of early diagnostics and vaccines; there are many commercially available tests for JD each with their own advantages and limitations. Most tests perform well at the herd level. But for the identification of individually infected animal, a combination of complementary tests, and repeated sampling can increase the diagnostic sensitivity for efficient control programs.
Diagnosis of the Map is a challenge due to the chronic nature and prevalence of stages of disease. Currently practiced serological testing for JD in animals includes agar gel immunodiffusion (Sherman et al., 1984) and commercial ELISA tests are generally prefereed. The intradermal Johnin test and interferon gamma assays (IFN-ã) (Wood et al, 1991, Rothel et al., 1992) are CMI based tests and the results are not reliable because of using the crude antigens which u showed variability in potency and cross reactivates. The bacterial isolation from feacal and tissue samples is considered as a gold standard for definitive diagnosis, though consuming.
Since the sequencing and analysis of the entire Map genome it has become possible to identify several unique ORF’s. These unique ORF’s on expression in heterologous E. coli system can be used to generate proteins for specific detection of Map in infected animals. In the present study the heterologous expression, purification and characterization of 843 bp unique ORF encoding 30.7 kDa protein of M. a. paratuberculosis has been studied.
Bacterial Strains and Plasmid
M. a. paratuberculosis strain 316F was obtained from Central Diengenees Kundig Tieh Institute, Lelystad, The Netherlands, and maintained at Gene Expression Laboratory, Veterinary Biotechnology Division, IVRI, Izatnagar, India. The mycobacteria were grown and maintained at 37æ%C on Middlebrook 7H10 agar (Difco laboratories, Detroit, USA) enriched with 0.1% glycerol (v/v) and 10% oleic acid dextrose catalase (Difco laboratories and supplemented with of 20 mg/L mycobactin J (AlliedMonitor, Fayette, USA) was also included. E. coli strain (M15, pREP4) supplied by Qiagen (Valencia, USA) was grown at 37æ%C in Luria Bertani broth containing kanamycin (25 µg/mL), as the strain carries kanamycin resistant plasmid. The expression vector plasmid pQE30 was purchased from Qiagen. The plasmid contains a T5 promoter and a 6 X His-tag coding sequence at 5’ to the multiple cloning region. The plasmid also contains an ampicillin resistance marker.
Oligonucleotide primers
To amplify a 17 kDa protein of Map, the primers: Map17F (sense) 5’ ) 5’ TAC GAGCTCATG CCG TCG CTG AAG CTC AC -3’29 mer and Map17R (antisense)5’ CGG AACCTT TCA GTCGATCCGGTCG -3’-30 mer, respectively, containing SacI/HindIII restriction endonuclease sites were designed on the basis of sequence information of Map str. k10, complete genome Gene Bank Accession No. AE016958 tag Map 3437c region 3816702-3817544.
Preparation of DNA
The genomic DNA from M. a. paratuberculosis was isolated from the grown culture by the method of Portillo et al. (Portillo et al., 1991). Plasmid DNA extraction from E. coli was carried out by the alkaline lysis method (Sambrook et al., 2001).
PCR Amplification and Cloning
The amplification reaction was performed in a 25 µL reaction volume containing 100 ng of M. a. paratuberculosis DNA; 2.5 µL of Taq DNA polymerase buffer 10 mM/L Tris.HCl (pH 9.0), 50 mM/L KCl, 1.5mM/L MgCl2 and 0.01% (w/v) gelatin; 200 µM of each dNTP; 0.5 µM each primers; 1 units of Taq DNA polymerase. The final volume was made up with sterile distilled water. The reaction was carried out in a PTC-100 reactor
(MJ Research Inc., Waltham, USA) for 30 cycles each cycle consisting of denaturation at 94æ%C for 1 minute, annealing at 55æ%C for 1 minute, and extension at 72æ%C for 1 minute. The amplified product was analyzed by submarine gel electrophoresis (Genei, Bangalore, India) on 1% agarose gel. The amplified gene productwas purified fromagarose gel using a QIAEXII gel extraction kit (Qiagen). For the purpose of cloning, about 500 ng of the PCR product was ligated into the plasmid pQE30 expression vector. The resulting plasmid p30.7 was transformed into competent E. coli M15 cells. The recombinant clones were selected on LB agar containing ampicillin (75 µg/mL) and kanamycin (25 µg/mL).
Transformants were further screened by restriction enzyme analysis of the plasmids with Sac I and Hind III (Sambrook et al., 2001). pUC18 Sau 3AI/TaqIdigest were used as standard molecular weight marker for agarose gel electrophoresis (Genei).
Expression and Purification of the Recombinant 30.7 kDa His-Fusion Protein
E. coli M 15 cells harbouring the plasmid pQ30.7 were grown in LB medium containing (75 µg/mL) and kanamycin (25 µg/mL) and induced with 1.0mM IPTG for 4–6 hours. The purification of recombinant 30.7 kDa His fusion protein under denaturing conditions was carried out by single step affinity chromatography using Ni-NTA (nickel-nitrilotriacetate) agarose (Qiagen) and renatured as described previously (Basagoudanavar et al., 2004). The protein concentration was determined spectrophotometrically (Lowry et al., 1951).The protein solution was sterilized by filtration, and aliquots were stored at 70°C, until used.
Two New Zealand White rabbits (8–10 weeks old) obtained from Laboratory Animals Resource section, IVRI, Izatnagar were used to raise antibodyagainst the recombinant 30.7 kDa His-fusion protein. Rabbits were immunized subcutaneously with 150 µg of immunogen with incomplete Freund’s adjuvant (IFA) (Genei, Bangalore, India), and boosters of 100 µg of immunogens with IFA were given intramuscularly after 3weeks and again 2 weeks later. Animals were bled ten days after the second booster and sera samples separated and stored at –20°C. All the sera used in this study were preabsorbed with E. coli antigens following the procedure of Harlow and Lane (Laemmli et al., 1970). In brief, recombinant 30.7 kDa His-fusion protein was initially electrophoresed and transferred to nitrocellulose membrane. The specific protein band was excised from the blot and incubated with polyclonal rabbit serum diluted 1 : 3 in buffer I (0.1M NaH2PO4, 0.01M Tris.Cl, pH 7.5) at 37°C, overnight. Following washing the membrane in buffer I, the monospecific antibodies were eluted in buffer II (0.1M NaH2PO4, 0.01M Tris.Cl, pH 6.3). These were utilized in western blotting experiments.
SDS-PAGE and Western Blotting
SDS-PAGE of the expressed recombinant 30.7 kDa His-fusion protein was carried out on a vertical slabmini apparatus (Atto, Tokyo, Japan) using polyacrylamide gels run under denaturing conditions as described by Laemmli in (Towbin et al., 1979). The 12% separating and 4% stacking polyacrylamide gel contained 0.1% SDS. About 25 µg of each sample was boiled in equal volume of 2X sample loading buffer prior to loading. Electrophoresis was carried with Tris-glycine electrode buffer (1X), pH 8.3 at 90Vfor 2 hours; the gels were stained overnight with Coomassie brilliant blue G 250 (Sigma, Milwaukee, USA) (Bjerrum et al., 1986). The coloured protein marker range from 17 to 175 kDa (New England Biolab, USA) was used as size markers. Samples electrophoresed on 12% SDS-PAGE were transferred to nitrocellulose membranes (0.45 µM) using semidry electroblotting (Atto, Tokyo, Japan) at 0.8mA/cm2, following the method of Bjerrum and Schaffer-Nielsen (Kyte et al., 1982). The blots were blocked with 2% skimmed milk powder in PBS-T buffer (PBS containing 0.1% Tween-20) for 2 hours at room temperature. After washing with PBS-T buffer three times, the membranes were incubated for 2 hours at 37æ%C with antisera against 30.7kDa His-fusion protein (1: 10000 in PBS) raised in rabbit. Following further washing, the blots were incubated with a 1: 1000 dilution of HRP-labelled goat anti-rabbit IgG (Sigma) for 1 hour. After washing, the blot was dipped in substrate solution (0.02% diamino benzidine suspended in PBS containing 0.03% hydrogen peroxide in PBS pH 7.4) for a minute, till brown colour developed.
Nucleotide Sequencing and Deduced Amino Acid Analysis
The nucleotide sequence determined of the 843 bp gene encoding 30.7 protein of Map strain 316F has been deposited in nucleotide database. The deduced amino acid sequence of gene encoding30.7 kDa protein from Map was analyzed for hydrophobic domains according to Kyte and Doolittle algorithm (Li et al., 2005) using Lasergene software (DNASTAR, Madison,USA). The homology prediction was also done with the help of public server UNIPROT and PSI-BLAST.
PCR Amplification and Construction of Recombinant pQ30.7
A PCR product of 858 bp (843bp gene and 15 bp linker) was obtained on amplification at 55°C.Restriction digestion of the recombinant plasmid pQ30.7 with SacI and HindIII released an identical size fragment as seen on a 1% agarose gel (Figure 1).
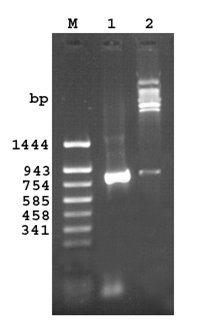
Fig 1. Agarose gel (1.2%) of the cloned fragment encoding 30.7 kDa protein of M. a. paratuberculosis
Lane M: DNA molecular weight marker pUC18 Sau 3AI/TaqIdigest
Lane 1: PCR amplified fragment encoding 30.7 kDa protein
Lane 2: Released insert of 843bp after SacI and Hind III digestion
Expression of Recombinant 30.7 kDa Protein in E. coli
When E. coli M15 cells harbouring the recombinant plasmid pQ30.7 were induced for 6 hours with 1 mM/L IPTG and analysed by 12% SDS-PAGE and Coomassie staining, a predominant band corresponding to that predicted for 30.7 kDa protein was detected in total cell extractof the E. coli (Figure 2, lane 2). The recombinant protein formed a major (more than 30%) portion of the total E.coli extract. No such protein band was observed with E.coli M15 cells or in uninduced E. coli M15 cells harbouring recombinant plasmid pQ30.7 (Figure 2, Lanes 3 and 4).
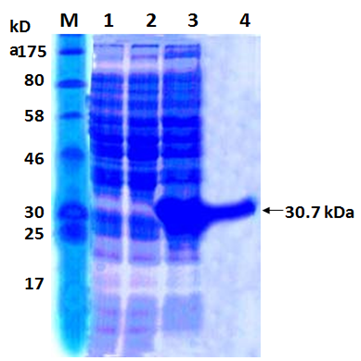
Fig. 2. SDS PAGE analysis of the recombinant 17 kDa protein
Lane M: Protein Marker
Lane 1: Cell extract of E. coli M15
Lane 2: Cell extract of E. coli M15 harbouring pQ30.7 clone un-induced
Lane 3: Cell extract of E. coli M15 harbouring pQ30.7 clone induced
Lane 4: Purified 30.7 kDa protein
Purification of the Recombinant 30.7 kDa Protein
The His-tagged purified protein, when analyzed on SDS-PAGE, showed a monomeric band of about 30.7 kDa size (Figure 2, Lane 1). Purification of the recombinant protein was nearly 80%, as visualized on SDS-PAGE. The yield of the pure recombinant protein was about 10–15 mg/L of culture at shake flask level.
Immunoreactivity of the Recombinant Protein
The polyclonal antiserum raised against purified 30.7 kDa protein in rabbits could bind to the IPTG that induced whole cell extract expressing recombinant as well as purified 30.7 kDa protein on western blot (Figure 3, Lanes 2 and 1). However no such band was visible in uninduced total cell extract of M15 harbouring pQ30.7 plasmid (Figure 3, Lane 3)
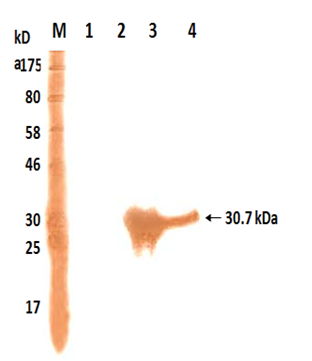 Fig. 3. Western Blot analysis of the recombinant 17 kDa protein
Fig. 3. Western Blot analysis of the recombinant 17 kDa protein
Lane M: Protein Marker
Lane 1: Cell extract of E. coli M15
Lane 2: Cell extract of E. coli M15 harbouring pQ17 clone un-induced
Lane 3: Cell extract of E. coli M15 harbouring pQ17 clone induced
Lane 4: Purified 17 kDa protein
Deduced Amino Acid Analysis
The nucleotide sequence of the pQ 30.7 plasmid having 843 bp gene of Map has been deposited in Gene bank database under accession no. EF 210209. The Blast search of the sequence revealed that no other mycobacterial sequences have homology. The predicted 281 amino acids of the 843 bp gene fragment had a mature are protein of 30.7kDa. Analysis of the deduced 281 amino acids sequence indicated that the protein contained four major hydrophobic regions (amino acids 18–25,119-126, 217-224 and 264-271)
JD caused by M.a. paratuberculosis, with its significant impact on livestock economy worldwide, has become a disease of concern. Currently, all antigen based diagnostic tests for JD use a complex mixtures of antigens, many of which are highly conserved among mycobacterial species. Diagnostic antigens appear to show promise, but none has been proved useful as a routine diagnostic tool.
The study of the completn genome of the M. a. paratuberculosis has accelerated the understanding the biology of this significant animal pathogen and also provide information about several ORFs specific only to pathogen (Leroy et al., 2009). Analysis of the M. a. paratuberculosis genome has revealed a total of 87 predicted coding sequences that are not present in available sequence databases, suggesting that these genes are unique and specific to M. a. paratuberculosis (Skeiky et al., 2000).
The next critical step will be learning functional roles of all the protein encoded by the ORFs. An achievement of this objective depends on the rapid expression and purification of proteins on a large scale. Therefore, the present study was envisaged to address the critical importance of identifying species specific antigens through the cloning and heterologous expression of unique predicted coding sequence found in the M. a. paratuberculosis genome.
An unique sequence of 843 bp from the Map genome with no homology with other mycobacteria and efficient expression system, based on the pQE30 vector, to produce 30.7kDa protein of Map in E.coli, so as to facilitate further characterization of the protein. Based on the sequence information of the gene encoding 30.7 kDa protein of Map str. k10, complete genome Gene Bank Accession No. AE016958 (tag Map 1636c region 1790543-1791016 coding for hypothetical protein) and also the information on the multiple cloning site of the expression vector pQE30 vector, restriction sites for SacI/HindIII were incorporated into the primers to facilitate directional cloning. In the design of the forward primer, consideration was given to inserting the amplified gene of 30.7kDa protein in frame into the pQE30 vector under the T5 promoter. The resulting plasmid pQ30.7 contained an open reading frame encoding successively 6X.His polypeptide and the 30.7kDa protein. The recombinant pQ30.7 clones were confirmed by release of the insert by double digestion with SacI/HindIII enzymes and on induction with IPTG appearance of the 17kDa band consistent with the predicted size of 30.7kDa protein. A similar strategy of cloning in pQE series expression vector system was applied to generate high levels of 35 kDa protein of Map (Basagoudanavar et al., 2004) and a 26kDa protein of Brucella abortus (Sisk et al., 1994). Expression of recombinant proteins is induced by, IPTG which binds to the lac repressor protein, inactivating it leading to transcription of sequences downstream of the promoter Expression of the gene facilitated the production of large amounts of the recombinant protein for immunological studies. The presence of 6X-histidine tag is of 840 Da in size at N-terminal of the recombinant protein facilitated single step affinity purification. It rarely interferes with protein immunogenicity, protein functional structure, hence the tag was not removed by protease cleavage (Sisk et al., 1994).
Authenticity of the purified recombinant protein was confirmed by reaction of the protein with the antisera raised against the protein. The antisera reacted strongly against purified 30.7 kDa as well as recombinant 30.7 kDa expressed in E. coli lysate in western blot showing their specificity and antigenicity.
The analysis of the deduced amino acid sequence according to the Kyte and Doolittle algorithm (Kumar et al., 2008), showed the presence of four highly hydrophobic regions, which could be the membrane segment of the protein. The homology study and deduced amino acid sequence are unique to Map and have the potential to be developed as diagnostic reagent.
The characterization of M. a. paratuberculosis specific ORF was one of the major challenge that has been addressed in this work. The high level expression of recombinant 30.7 kDa provides opportunity for its further characterization. The results of this study suggested that a specific protein 30.7kDa could be useful for the development of diagnostic reagent. Extensive screening with sera of clinical and subclinically affected cases of paratuberculosis are needed to ascertain the diagnostic potential with regard to specificity and sensitivity with 30.7kDa as antigen.
ACKNOWLEDGMENTS
The authors are highly thankful to Director, Indian Veterinary Research Institute, India for providing necessary facilities for conducting the research.
- Basagoudanavar, S.H., Goswami, P.P., Tiwari, V. Pandey, A.K. and Singh, N., Heterologous expression of a gene encoding a 35kDa protein of Mycobacterium avium paratuberculosis in Escherichia coli. Vet. Res. Commun., 2004; 28: 209–224.
- Bjerrum, O.J., and Schafter-Nielson, C., Buffer systems and transfer parameters for semidry electroblotting with a horizontal apparatus. Electrophoresis 86, Dunn, Ed., 1986; 315-327, VCH,Weinheim, Germany.
- Clarke, C.J., The pathology and pathogenesis of paratuberculosis in ruminants and other species. J. Comp. Pathol., 1997; 116: 217–261.
- Gangadhararao A., Das, D., Veerasami, M., Senthilkumar, R.L., Durishetty, M., Ramalakshmi, B., Bahekar, V., Mukherjee, F., Chandran, D., Kumar, P.U., Sesikeran B. and Srinivasan, V.A., Antemortem and postmortem examinations of the cattle calf naturally infected with Mycobacterium avium subsp. paratuberculosis Euro. J. Microbiol. Immunol., 2013; 4: 241-251.
- Harlow, E., and Lane, D., Antibodies, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA, 1988.
- Kumar, S., Tuteja, U., Kumar, A., and Batra, H.V., Expression and purification of the 26 kDa periplasmic protein of Brucella abortus: a reagent for the diagnosis of bovine brucellosis, Biotechnol. Appl. Bioc., 2008; 49: 213–218.
- Kyte, J., and Doolittle, R.F., A simple method for displaying the hydropathic character of a protein. J.Mol., Biol., 1982; 157: 105–132.
- Laemmli, U.K., Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature, 1970; 227: 5259 680–685.
- Leroy, B., Viart, S., Trinchero, N., Roupie, V. and Govaerts, M., Use of Mycobacterium avium subsp. paratuberculosis specic coding sequences for serodiagnosis of bovine paratuberculosis.Vet. Microbiol., 2009; 135: 313-319.
- Li, L., Bannantine, J.P., Zhang, Q., Amonsin, A., May, B.J., Alt, D., Banerji, N., Kanjilal, S. and Kapur, V., The complete genome sequence of Mycobacterium avium subspecies paratuberculosis. P. Natl. Acad. Sci-Biol.,2005; 102: 12344–12349.
- Lowry, O.H., Rosebrough, N.J., Farr, A.L., and Randall, R.J., Protein measurement with the Folin phenol reagent. J Biol. Chem., 1951; 193: 265–275.
- Ott, SL., Wells, SJ. And Wagner, BA., Herd-level economic losses associated with Johne’s disease on US dairy operations. Prev. Vet. Med., 1999; 40: 179–192.
- Portillo, P. D. Murillo, L. A., and Patarroyo, M. E., Genetically engineered vaccines: an overview Plasmid., 1991; 39: 100–113.
- Rothel, JS., Jones, SL., Corner, LA., Cox, JC. And Wood, PR. The gamma-interferon assay for diagnosis of bovine tuberculosis in cattle: conditions affecting the production of ammainterferon in whole blood culture. Aus. Vet. J., 1992; 69: 1–4.
- Roupie, V., Viart, S., Leroy, B., Romano, M., Trinchero, N., Govaerts, M., Letesson, JJ., Wattiez, R. and Huygen, K., Immunogenicity of eight Mycobacterium avium subsp. paratuberculosis specific antigens in DNA vaccinated and Map infected mice. Vet. Immunol. Immunop., 2012; 145: 74–85.
- Sambrook, J., and Russel, D.W., Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, Cold Spring Harbor, New York, USA, 3rd edition, 2001.
- Sherman, DM., Markham, RJ. and Bates, F., Agar gel immunodiffusion test for diagnosis of clinical paratuberculosis in cattle. J. Am. Med. Assoc., 1984; 185: 179–182.
- Sisk, W.P., Bradley, J.D. and Leipold, R.J. High-level expression and purification of secreted forms of herpes simplex virus type 1 glycoprotein gD synthesized by baculovirus-infected insect cells, J. Virol., 1994; 68: 766–775.
- Skeiky, Y.A.W., Pamela, J., and Ovendale, P.J., T cell expression cloning of a Mycobacterium tuberculosis gene encoding a protective antigen associated with the early control of infection. J. Immunol., 2000; 165: 7140–7149.
- Stehman, SM. Paratuberculosis in small ruminants, deer,and South American camelids. Vet. Clin. N. Am-Food. A., 1996; 12: 441–455.
- Stehman, S.M. Paratuberculosis in small ruminants, deer,and South American camelids. Veterinary Clinics of North America: Food Animal Practice 1996; 12, 441–455 .
- Taylor, T.K., Wilks, C.R. and McQueen, D.S. Isolation of mycobacteriumparatuberculosis from the milk of a cow with Johne’s disease. Vet. Rec., 1981; 109: 532–533.
- Towbin, H., Staehelin, T., and Gordon, J., Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. P. Natl. Acad. Sci-Biol., 1979; 76: 4350–4354.
- Wood, P.R., Corner, L.A., Rothel, J.S., Baldock, C., Jones, S.L., Cousins, D.B., McCormick, B.S., Francis, B.R., Creeper, J., Tweddle, N.E. Field comparison of the interferon-gamma assay and the intradermal tuberculin test for the diagnosis of bovine tuberculosis. Aus. Vet. J., 1991; 68: 286–290.
© The Author(s) 2016. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.