


ISSN: 0973-7510
E-ISSN: 2581-690X
Classical Swine Fever (CSF) remains a highly transmissible viral threat to the Suidae family with considerable economic consequences worldwide. The current study developed and comprehensively validated an indirect ELISA that uses the recombinant E2 protein (rE2) from CSFV. The amplified 546 bp segment from the N-terminal of the CSFV E2 gene, cloned into the pET-28a vector, and introduced into E. coli TOP10 and subsequently into BL21(DE3) cells. A ~25 kDa His-tagged rE2 protein was successfully expressed, followed by solubility analysis and purified using affinity chromatography. The recombinant protein was verified by Western blot analysis employing anti-His antibodies and CSFV positive pig sera. Using 500 pig serum samples, the developed iELISA diagnostic performance was evaluated and compared with both an indigenously developed Erns ELISA kit and Priocheck CSFV Antibody ELISA kit. ROC analysis of developed rE2 iELISA demonstrated robust diagnostic precision, yielding an area under the curve (AUC) value of 0.935 (92% sensitivity, 90% specificity; cut-off 0.5695) against the Priocheck CSFV Antibody ELISA kit and with indigenously developed Erns ELISA assay (AUC 0.995; 97.98% sensitivity; 96.97% specificity at the cut-off of 0.404). A statistically significant variation in diagnostic accuracy of two assays was evident in comparative ROC curve analysis (AUC difference = 0.216; SE = 0.0482; 95% CI: 0.122-0.311; z = 4.484; P < 0.0001) proving the developed assay provides a sensitive, specific, and robust serological tool suitable for CSF serosurveillance.
Classical Swine Fever Virus (CSFV), Recombinant E2 Protein (rE2 Protein), Indirect Enzyme Linked Immuno Sorbent Assay (iELISA), Serological Diagnosis, Diagnostic sensitivity and specificity, ROC Analysis, Immunomonitoring
Classical Swine Fever (CSF), historically referred to as hog cholera, remains a critical viral disease affecting the global swine industry attributable to its high morbidity rates and the rapid spread among domestic swine herds and feral boar cohorts. The disease imposes considerable economic burdens, including production losses, trade restrictions, and expenditures incurred during eradication.1,2 The etiological agent, Classical Swine Fever Virus (CSFV), is an enveloped, positive-sense, single-stranded RNA virus classified within the genus Pestivirus, family Flaviviridae.3 The approximately 12.3 kb genome encodes a single large polyprotein, which is subsequently cleaved co- and post-translationally into structural (C, Erns, E1, E2) and non-structural proteins (Npro, p7, NS2-NS5B).4 Of these, the E2 glycoprotein functions as the principal immunodominant antigen, eliciting virus-neutralizing antibodies and playing a pivotal role in viral attachment and the induction of protective immunity. Due to its high immunogenicity and relative sequence conservation, E2 is widely acknowledged as a prime target for serological assays and vaccine development.5 Notably, the B-cell antigenic domains (BCADs) within the E2 protein are essential for antibody recognition, making them valuable elements in the design of immunoassays.
Accurate and rapid detection of CSFV is essential for effective disease surveillance, outbreak management, and long-term prevention strategies. This is especially important in regions where the disease remains endemic or eradication campaigns are ongoing.6,7 In recent years, several laboratory diagnostic approaches have been established. These encompass propagation of virus in cell culture, fluorescent antibody assays, antigen-capture ELISA, nucleic acid amplification techniques such as Reverse Transcriptase-Polymerase Chain Reaction (RT-PCR) real-time PCR, a range of serological tests.8,9 Among these, the Enzyme-Linked Immunosorbent Assay (ELISA) has emerged as one of the most extensively adopted diagnostic tools because of its operational simplicity, high throughput capacity, affordability, and suitability for both laboratory and field surveillance contribute to its popularity.10 Importantly, ELISA-based detection supports DIVA (Differentiating Infected from Vaccinated Animals) strategies and this approach is vital for maintaining disease-free status and implementing movement controls in swine populations.11
Although early ELISA techniques offered certain advantages, those utilizing crude cellular extracts or whole-virus antigens frequently exhibited cross-reactivity with antigenically related pestiviruses, including Border Disease Virus (BDV) and Bovine Viral Diarrhea Virus (BVDV).6,12 In regions with prevalent mixed pestivirus infections, this antigenic overlap often led to reduced diagnostic specificity and an increased incidence of false positive results, thereby complicating CSF surveillance.13 To mitigate these challenges, research efforts have increasingly concentrated on recombinant protein-based ELISAs, particularly those employing truncated or domain-specific segments of the E2 glycoprotein, such as B-cell antigenic domains (BCADs). These modified antigen constructs reduce cross-reactive epitopes while preserving immunodominant regions necessary for antibody detection, thereby enhancing diagnostic specificity.7,14
Antibody detection is now significantly more accurate and useful owing to recent developments in ELISA technology. The sensitivity and specificity of these assays have been augmented by the advancement of blocking and double-antigen ELISAs, competitive ELISAs employing nanobodies, and recombinant truncated E2 constructs. Additionally, technological developments such as digital ELISA, microarray-based ELISA, and nanoparticle-enhanced detection methods have enabled increased sensitivity and specificity, enabling rapid and quantitative antibody detection.9,15,16 Recombinant E2-based ELISA has emerged as a reliable and efficient technique for identifying CSFV antibodies in both laboratory and field samples; therefore, the present study was undertaken to develop an rE2-based indirect ELISA.
Primer design and amplification of BCAD domain of E2 glycoprotein
The B-cell antigenic domain (BCAD) region of the E2 gene was selected as the amplification target. The nucleotide sequence of the E2 gene of CSFV was retrieved from the National Center for Biotechnology Information (NCBI) database (Accession No. KM262189 and MH734359). Primers were designed using NCBI Primer-BLAST and Integrated DNA Technologies (IDT) tools to specifically amplify the BCAD domain (~2720-3320 nt). To facilitate directional cloning into the pET-28a expression vector, recognition sequences for the restriction enzymes BamHI and XhoI were inserted at the 5′ ends of the forward and reverse primers, respectively (Table).
Table. Details of Designed Primers
Target gene |
BCAD region of E2 gene |
|---|---|
Forward Primer |
CGCGGATCCCGGTTAGCCTGCAAGGA |
Reverse Primer |
GTGCTCGAGTTCTGCAAGGTAATCT |
RT-PCR amplification was performed (Bio-Rad, S1000 Thermal cycler) in a 25 μl reaction volume using TAKARA kit (Cat. No. RR057A). Each reaction mixture contained 12.5 μl of 2X buffer containing dNTP mix, 1 μl of enzyme mix, 1 μl each of forward and reverse primers, 3.5 μl of nuclease-free water, 1 μl of DMSO, and 5 μl of template RNA isolated from the supernatant of PK-15 cells infected with CSFV field strain (genotype 1.1). The cycling conditions were as follows: cDNA synthesis (reverse transcription) at 50 °C for 30 minutes; initial denaturation at 94 °C for 2 minutes; 30 cycles of denaturation at 94 °C for 30 seconds, annealing at 56.2 °C for 30 seconds, and extension at 72 °C for 30 seconds; final extension at 72 °C for 5 minutes; and a hold at 4 °C. PCR products were analyzed by 1.2% agarose gel electrophoresis. The expected amplicon was visualized under UV illumination (Vilber Lourmat, Bio-Print CX4) and gel-purified using the gel purification kit (MN, Cat. No. 740609.50).
Cloning and expression of E2 gene
The gel-purified amplicon and pET-28a vector were enzymatically digested with BamHI and XhoI restriction enzymes at 37 °C for 3 hours, followed by heat inactivation and gel purification. The digested insert was ligated into the linearized pET-28a vector using T4 DNA ligase (Thermo Scientific Cat. No. EL0011) at a 3:1 molar ratio of insert to vector, with the ligation mixture incubated overnight at 4 °C. The ligated product was introduced into E. coli TOP10 competent cells via the heat-shock method. Positive transformants were selected on LB agar plates supplemented with kanamycin (50 μg/ml) and subsequently verified by colony PCR and restriction analysis.
Expression of recombinant E2 protein
The recombinant plasmid (pET-28a-rE2-BCAD) was incorporated into E. coli BL21(DE3) cells to facilitate protein expression. A single positive colony was transferred to LB broth containing kanamycin (50 μg/ml) and chloramphenicol (35 μg/ml) and incubated overnight at 37 °C with shaking at 180 rpm. The overnight culture was diluted 1:100 into fresh LB broth containing the same antibiotics and incubated at 37 °C until the optical density (OD) reaches 0.4-0.6. The expression of recombinant protein was upregulated by adding Isopropyl β-D-1-thiogalactopyranoside (IPTG) to a final concentration of 1 mM, followed by incubation at 37 °C for 5 hours to promote soluble protein production. Induced cells were collected by centrifugation at 6000 rpm for 15 minutes at 4 °C, and the resulting cell pellet was stored at -80 °C for further analysis.
Solubility test for expressed recombinant protein
About 1 ml aliquot of the induced E. coli culture was harvested by centrifugation, and the pellet was reconstituted in 100 μl of Bug-Buster reagent (Merck, Cat. No. 70584). The suspension was incubated at 37 °C for 30 minutes with gentle shaking at 90 rpm to allow the non-ionic detergents in Bug-Buster to permeabilize the E. coli cell walls while preserving protein functionality. Following incubation, the mixture was subjected to centrifugation to separate the soluble (supernatant) and insoluble (pellet) fractions. Both fractions were subjected to Sodium Dodecyl Sulfate–Polyacrylamide Gel Electrophoresis (SDS-PAGE) analysis to evaluate the solubility profile of the expressed rE2 protein.
Purification of rE2 protein by Ni-NTA column chromatography
The pelleted bacterial cells were washed with wash buffer and centrifuged at 10,000 rpm for 10 minutes at 4 °C. The pellet was redispersed in lysis buffer (10 mM imidazole, 100 mM NaH2PO4, 10 mM Tris base, 8 M urea, pH 8.0) and subjected to sequential physical (three freeze-thaw cycles), enzymatic (1 mM lysozyme), and mechanical sonication (Hielscher Ultrasonics, P400S ultrasonic processor) in an ice bath at 60% amplitude, 30 sec pulse-on and 30 sec pulse-off for 10 cycles] lysis methods to maximize cell disruption. The lysate was subsequently centrifuged at 15,000 rpm for 30 minutes at room temperature to separate the soluble fraction. The resulting supernatant, containing soluble recombinant protein, was subjected to a Ni-NTA column pre-equilibrated at pH 8.0 for affinity purification. The column was washed with wash buffer (20 mM imidazole, 100 mM NaH2PO4, 10 mM Tris base, 8 M urea, pH 6.0) to remove non-specifically bound proteins, and the His-tagged recombinant protein was eluted with elution buffer (300 mM imidazole, 100 mM NaH2PO4, 10 mM Tris base, 8 M urea, pH 6.0). Eluted fractions were assessed for protein concentration by measuring absorbance at 280 nm using a NanoDrop spectrophotometer, and pooled accordingly. The pooled fractions were subsequently dialyzed against dialysis buffer (100 mM NaH2PO4, 10 mM Tris base, pH 7.4) at 40 °C for removal of urea and imidazole. After dialysis the purified protein was stored at -80 °C for downstream applications.
SDS-PAGE analysis
Recombinant protein expression and purity were assessed by SDS-PAGE with a 10% resolving gel under denaturing conditions. Equal volumes of induced, uninduced, pellet, supernatant and purified protein samples were combined with 2X Laemmli loading buffer containing β-mercaptoethanol, then heated at 99 °C for 15 minutes. Samples were loaded onto the gel alongside a molecular weight marker and the protein bands were visualized after staining with Coomassie Brilliant Blue R-250, followed by destaining.
Western blot analysis
For immunoblot confirmation, rE2 protein was resolved by SDS-PAGE and blot transferred to nitrocellulose (NC) membrane using a semi-dry transfer system. Blocking was performed overnight at 4 °C using 5% skim milk dissolved in 1X Phosphate Buffered Saline containing 0.05% Tween-20 (PBST).
Two separate blots were prepared: one was incubated for 1 hour at 37 °C with anti-His tag monoclonal antibody (1:5000) (Bethyl Laboratories, Cat. No. A190-114P), and the other was incubated with CSFV specific porcine convalescent sera (1:50) under the same conditions.
Followed by washing with PBST; 1:10,000 diluted Horseradish Peroxidase (HRP)-conjugated anti-pig IgG secondary antibody (Sigma-Aldrich, Cat. No. A5670) was added and allowed to react for 1 hour at 37 °C. 3,3′-Diaminobenzidine (DAB) (Sigma Aldrich, Cat. No. D4418) substrate was added for the detection of recombinant protein reactivity.
Development of indirect ELISA using rE2 protein (rE2-iELISA)
A modified indirect ELISA was established utilizing the purified rE2 protein as the coating antigen. The analytical sensitivity of the rE2 iELISA was determined by checkerboard titration to find out the minimum detectable range of antigen concentration and optimal serum dilution.
The purified rE2 protein was diluted in carbonate-bicarbonate coating buffer (pH 9.6) and coated to 96-well ELISA plates (Nunc-Maxisorp, Thermo Scientific, Cat. No. 442404) at concentrations ranging from 25, 50, 100, 150, 200 and 250 ng/well, followed by incubation for 1 hour at 37 °C. Plates were washed three times with 1X PBS containing 0.05% Tween-20 and subsequently blocked with 5% skimmed milk in PBST for 1 hour at 37 °C to minimize non-specific binding. The control serum samples were tested ranging from 1:25, 1:50, 1:100, 1:150, 1:200 and 1:250 dilution with blocking buffer, added to the wells (100 μl/well), and incubated at 37 °C for 1 hour. After washing, anti-pig IgG conjugated with HRP was added at dilution of 1:10,000 according to manufacturer guidelines and plates were incubated for 1 hour at 37 °C. 3,3′,5,5′-Tetramethylbenzidine (TMB) (MP Biomedicals, Cat. No. 02152346-CF) substrate was utilized for color development, and the reaction was terminated with 0.5 N H₂SO₄ after color development. OD was measured at 450 nm using a microplate reader (Model EPOCH2NS-SN, Agilent Technologies, Inc.). The optimal concentrations of antigen, serum, and conjugate, which provided the highest balanced positive-to-negative (P/N) ratio, were selected as working conditions for the assay.
Analytical specificity
The analytical specificity of the assay was determined by testing positive serum samples of other diseases of swine including Bovine Viral Diarrhea (BVD) (n = 06), African Swine Fever (ASF) (n = 10), Foot and Mouth Disease (FMD) (n = 10) and Porcine Reproductive and Respiratory Syndrome (PRRS) (n = 10) using developed rE2 ELISA.
Serum samples
A total of 500 unvaccinated pig serum samples were procured from serum repository maintained at ICAR-National Institute of Veterinary Epidemiology and Disease Informatics for the present study. The samples collected from different districts of Karnataka, namely Bagalkote (n = 44), Bangalore Rural District (n = 13), Belagavi (n = 75), Chikkaballapura (n = 26), Dharwad (n = 18), Gadag (n = 7), Kodagu (n = 13), Koppal (n = 13), Mandya (n = 78), Ramanagara (n = 26), Tumkuru (n = 13), Udupi (n = 18), Vijayapura (n = 65) and Yadagiri (n = 91) were used. The serum samples were stored at -20 °C until further use.
Evaluation of field samples and comparison with gold standard assay
A total of 500 pig serum samples from the serum repository were evaluated using the developed indirect ELISA. These samples were concurrently analyzed using the Priocheck CSFV Antibody ELISA Kit (Thermo Scientific, Cat. No. 7610046) and indigenously developed Erns ELISA (ICAR-NIVEDI-2024-051),17,18 serving as the reference (gold standard) assays. The diagnostic performance of the developed assay was assessed by calculating sensitivity, specificity, and concordance with the commercial kit.
Receiver Operating Characteristic (ROC) analysis
To further evaluate the diagnostic accuracy of the developed ELISA, receiver operating characteristic (ROC) curve analysis was conducted using the results from 500 serum samples. The Priocheck CSFV Antibody ELISA was employed as the reference standard to classify samples as true positives or true negatives. ROC analysis was conducted using MedCalc software (Version 19.1) to determine the area under the curve (AUC), optimal cutoff OD value, sensitivity, and specificity of the developed assay. The classification threshold for test samples was established by selecting the cut-off value that maximized Youden’s index (J = sensitivity + specificity −1).19
Amplification of the BCAD domain of the E2 glycoprotein
Primers incorporating BamHI and XhoI restriction enzyme recognition sequences were engineered to specifically amplify the BCAD region of the E2 gene. PCR amplification yielded a distinct 546 bp fragment, and agarose gel electrophoresis revealed a single, well-defined band, substantiating the primer set’s high specificity and affirming the successful amplification of the intended target region.
Construction of recombinant pET-28a-rE2-BCAD plasmid
The purified amplicon and pET-28a vector were subjected to BamHI and XhoI digestion, producing a fragment of the anticipated size. Following ligation, the recombinant construct was introduced into E. coli TOP10 competent cells. Transformants obtained on LB-kanamycin plates were screened by colony PCR, and several clones yielded the anticipated 546 bp product. Plasmid DNA isolated from positive clones was further validated by restriction enzyme digestion, which released the insert at the predicted size, confirming correct cloning into the pET-28a backbone.
Transformation into expression host and insert confirmation
The verified recombinant plasmid isolated from TOP10 cells was subsequently transformed into E. coli BL21(DE3) cells for protein expression. Kanamycin-Chloramphenicol-resistant colonies were successfully obtained. Restriction digestion of plasmid DNA extracted from BL21(DE3) transformants yielded digestion profiles consistent with the expected recombinant construct, confirming the presence and integrity of the insert.
Expression of rE2 protein
E. coli BL21(DE3) cells containing the recombinant pET-28a-BCAD construct were grown overnight and subsequently inoculated into fresh LB broth and were incubated at 37 °C at 200 rpm for protein expression analysis. The induction was performed when the OD600 reached 0.6-0.8. Protein expression was triggered by the addition of IPTG (Final concentration 1 mM), and cultures were incubated to allow expression of the rE2 protein for 4-5 hours under the same conditions. SDS-PAGE analysis of induced samples revealed a prominent band at ~25 kDa, corresponding to the expected size of the rE2 protein (Figure 1).

Figure 1. IPTG-induced expression of recombinant protein
L1 – Protein Marker, L2 to L5 – induced protein from different colony culture, L6 – NC (uninduced protein)
Solubility analysis of expressed rE2 protein
To assess protein solubility, the induced culture was separated into soluble and insoluble fractions. SDS-PAGE analysis revealed that the 25 kDa rE2 protein was predominantly localized within the insoluble pellet fraction.
Purification of rE2 protein
The insoluble protein fraction was purified by first solubilizing the inclusion bodies using standard denaturation and refolding protocols, followed by Ni-NTA affinity chromatography. This process yielded eluted protein fractions of approximately 25 kDa. The purified protein was obtained at high purity and concentration (2.1 mg/ml from 300 ml of induced culture) and subsequently stored at -80 °C for downstream applications (Figure 2).
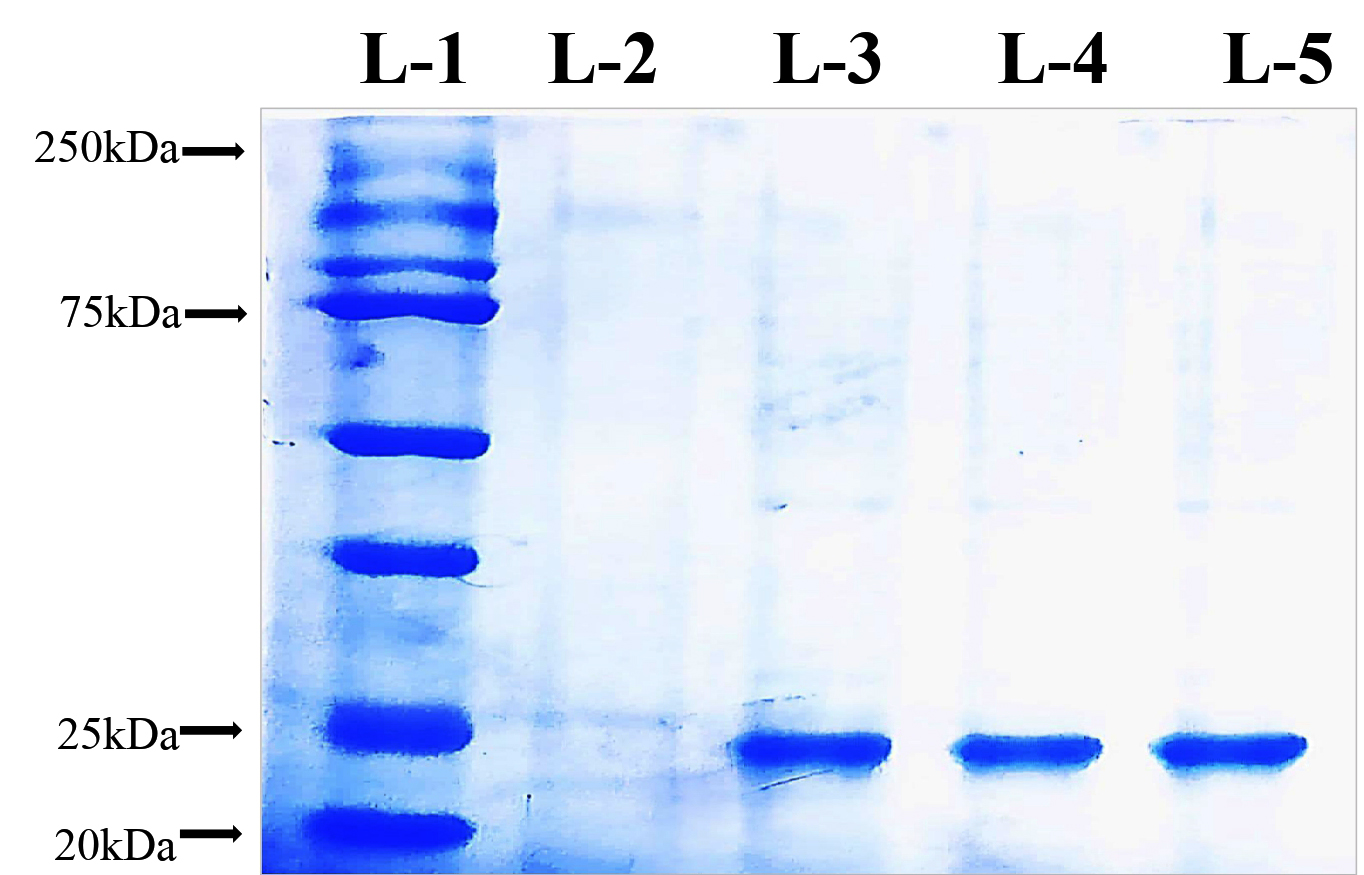
Figure 2. Purification of recombinant protein by affinity column chromatography
L1 – Protein Maker, L2 – Purified fraction from uninduced Protein, L3 to L5 – Purified rprotein Elution
Western blot confirmation of rE2 protein
Western blot analysis was conducted to validate the identity and immunoreactivity of the purified rE2 protein. The immunoblot revealed a distinct band at approximately 25 kDa when probed with an anti-His monoclonal antibody, confirming the presence of the His-tagged recombinant protein. Additionally, the same band exhibited strong reactivity with CSFV convalescent serum, indicating that the expressed protein retained relevant antigenic epitopes recognized by virus-specific antibodies (Figures 3 and 4).
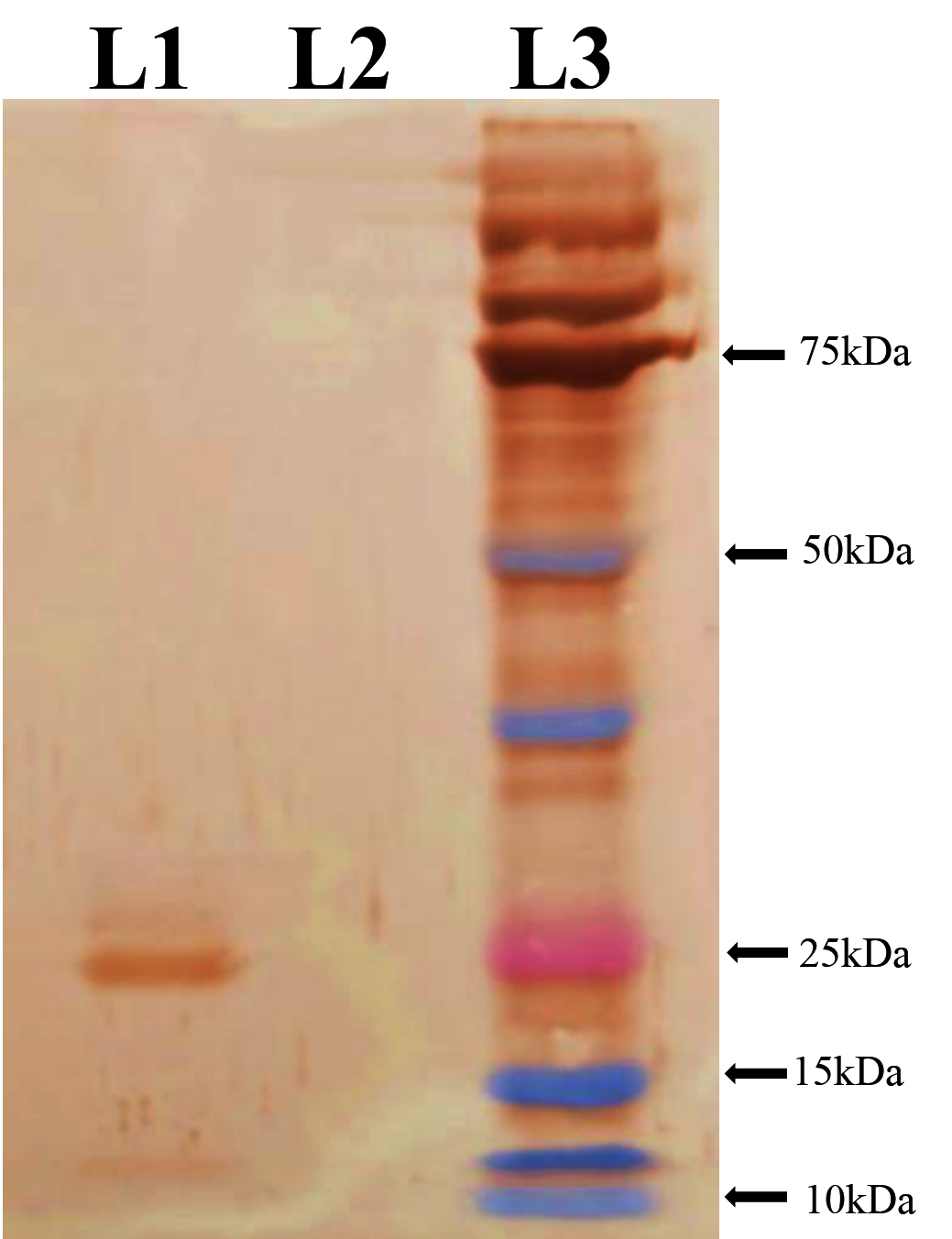
Figure 3. Immunoreactivity of recombinant protein with anti-His monoclonal antibody
L1 – rProtein, L2 – Uninduced Protein, L3 – Protein Maker
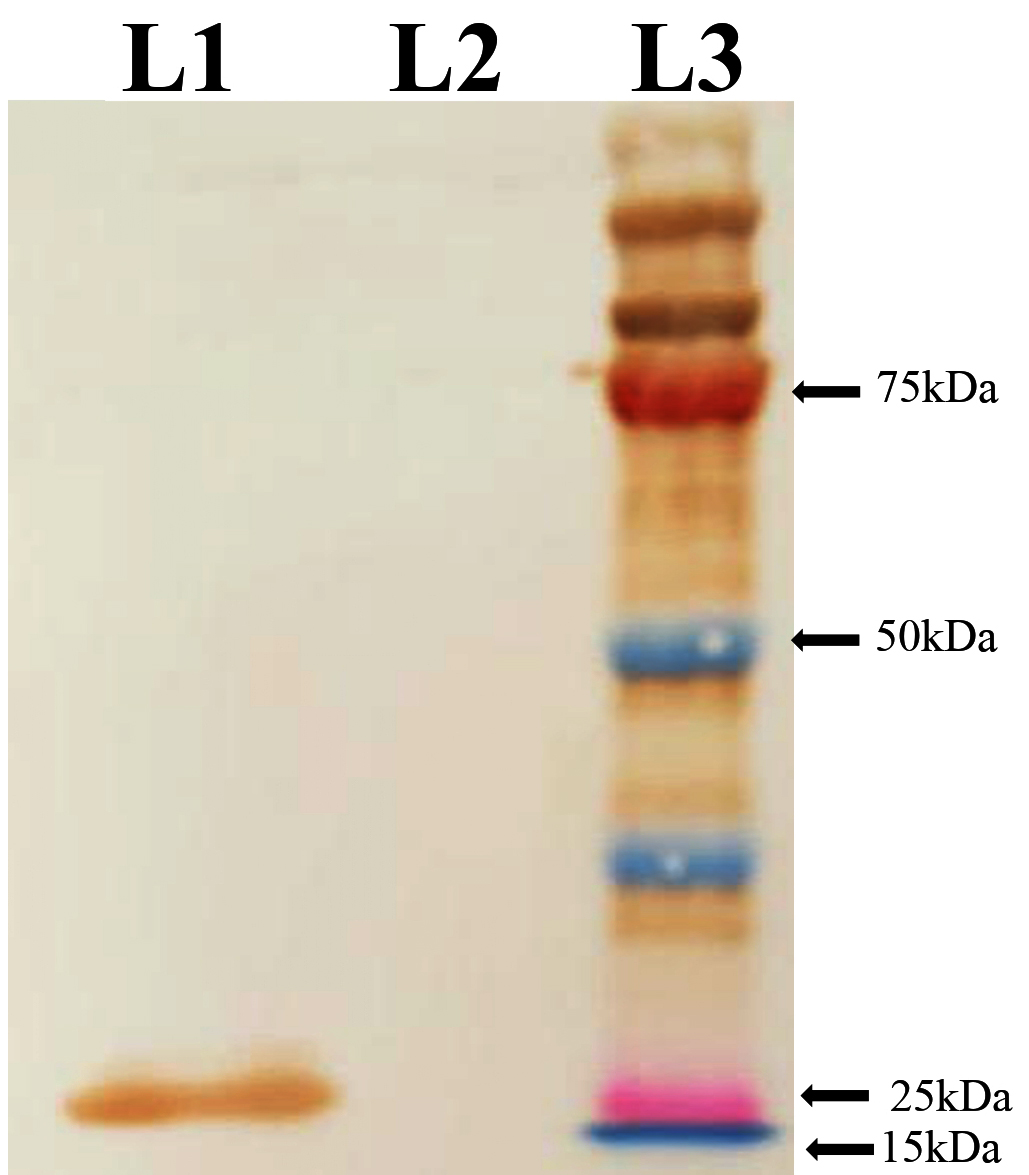
Figure 4. Reactivity of recombinant protein with CSFV convalescent pig sera
L1 – rProtein, L2 – Uninduced Protein, L3 – Protein Maker
Standardization of rE2-iELISA
An indirect ELISA was optimized using the purified rE2 protein as the coating antigen. The checker board titration was carried out to optimize the antigen and antibody concentration required for iELISA. Briefly, each well was coated with antigen at concentrations of 25, 50, 100, 150, 200 and 250 ng/well. Standard positive and negative serum samples (which were tested using Priocheck CSFV Antibody ELISA) were diluted from 1:25, 1:50, 1:100, 1:150, 1:200 and 1:250 at conjugate dilution of 1:10000. At all antigen concentrations, the OD values obtained with positive sera decreased gradually as serum dilution increased, showing an accurate dilution-dependent antibody response. Higher OD values observed at lower serum dilution (1:25 and 1:50) correlated with higher background reactivity and signal levels (Figure 5). The fold difference between positive and negative sera increased with antigen concentration up to 100 ng per well before receding. The highest fold difference (~5.2-fold) was observed at 100 ng/well at the working dilution of 1:100, whereas limited fold changes were observed at 25 ng (3.1-fold) and 50 ng (4.0-fold). Higher antigen concentrations (150-250 ng) continued to decline signal discrimination and consequently elevated background reactivity.
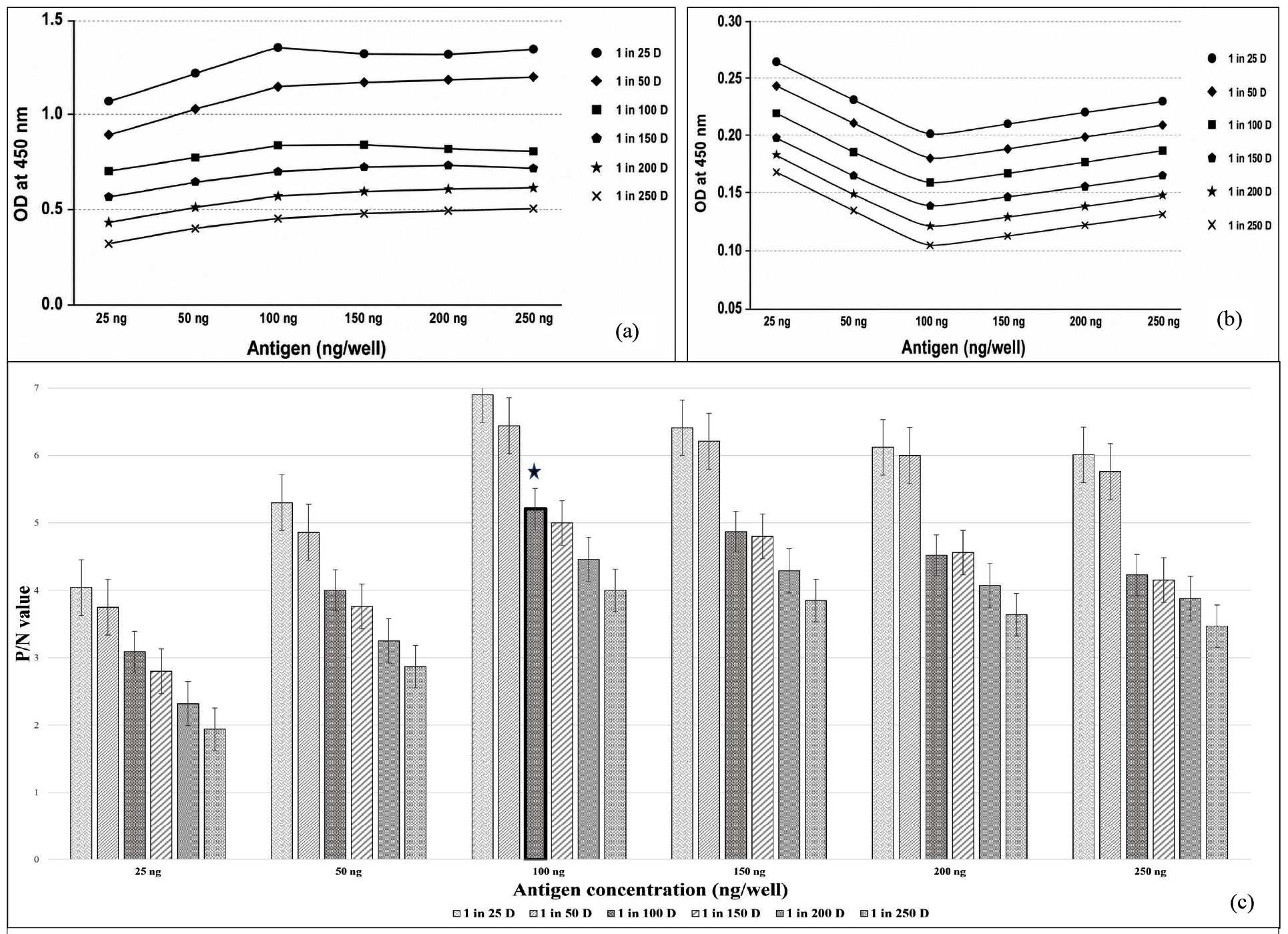
Figure 5. Optimization of indirect ELISA (iELISA) conditions by checkerboard titration.
(a) OD450 values of positive control serum tested at serial dilutions against graded antigen coating concentrations.
(b) OD450 values of negative control serum evaluated under identical antigen and serum dilution conditions to assess background reactivity.
(c) Positive-to-negative (P/N) ratio calculated for each antigen concentration and serum dilution combination
The checkerboard titration established an optimal antigen coating concentration of 100 ng per well. The primary antibody (serum) dilution was standardized at 1:100, while the secondary antibody (HRP-conjugated) provided the best signal-to-noise ratio at a dilution of 1:10,000. Blocking conditions were optimized by incubating the plates for one hour at 37 °C, which effectively minimized non-specific binding. All ensuing incubation phases were also performed for one hour at 37 °C, resulting in consistent and reproducible absorbance values.
Analytical specificity
Using rE2 protein as coating antigen to determine the cross reactivity with heterologous samples were assessed. No cross reactivity was observed with BVDV, ASFV, FMDV and PRRSV serum samples, indicating high specificity of the developed rE2 iELISA.
Comparison and cutoff calculation by ROC analysis
A receiver operating characteristic (ROC) curve analysis was conducted to validate the diagnostic performance and optimal cut-off value of the developed rE2-iELISA. When compared with the indigenously developed Erns ELISA, the assay demonstrated near-perfect accuracy, yielding an area under the curve (AUC) of 0.995. The optimal cut-off value corresponding to the maximum Youden index was 0.404, with sensitivity of 97.98% and specificity of 96.97% (Figure 6). Evaluation against the commercial Priocheck CSFV Antibody ELISA using the same sample set showed an AUC of 0.935, with 92% sensitivity and 90% specificity at a cut-off value of 0.5695 (Figure 7).
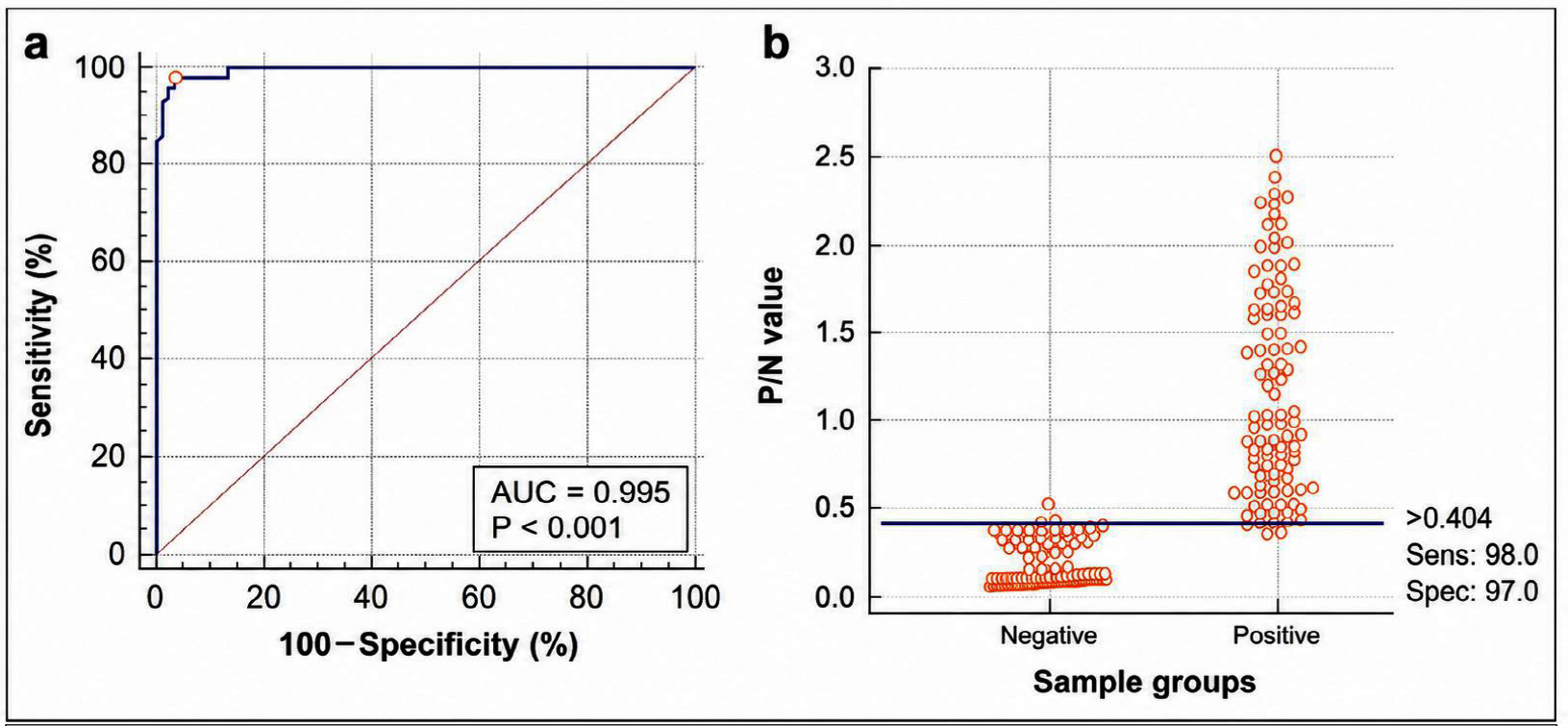
Figure 6. (a) Receiver Operating Characteristic (ROC) analysis of the developed rE2-iELISA against the NIVEDI in-house antibody detection kit. (b) Distribution of positive and negative serum samples based on P/N values with the selected cutoff point
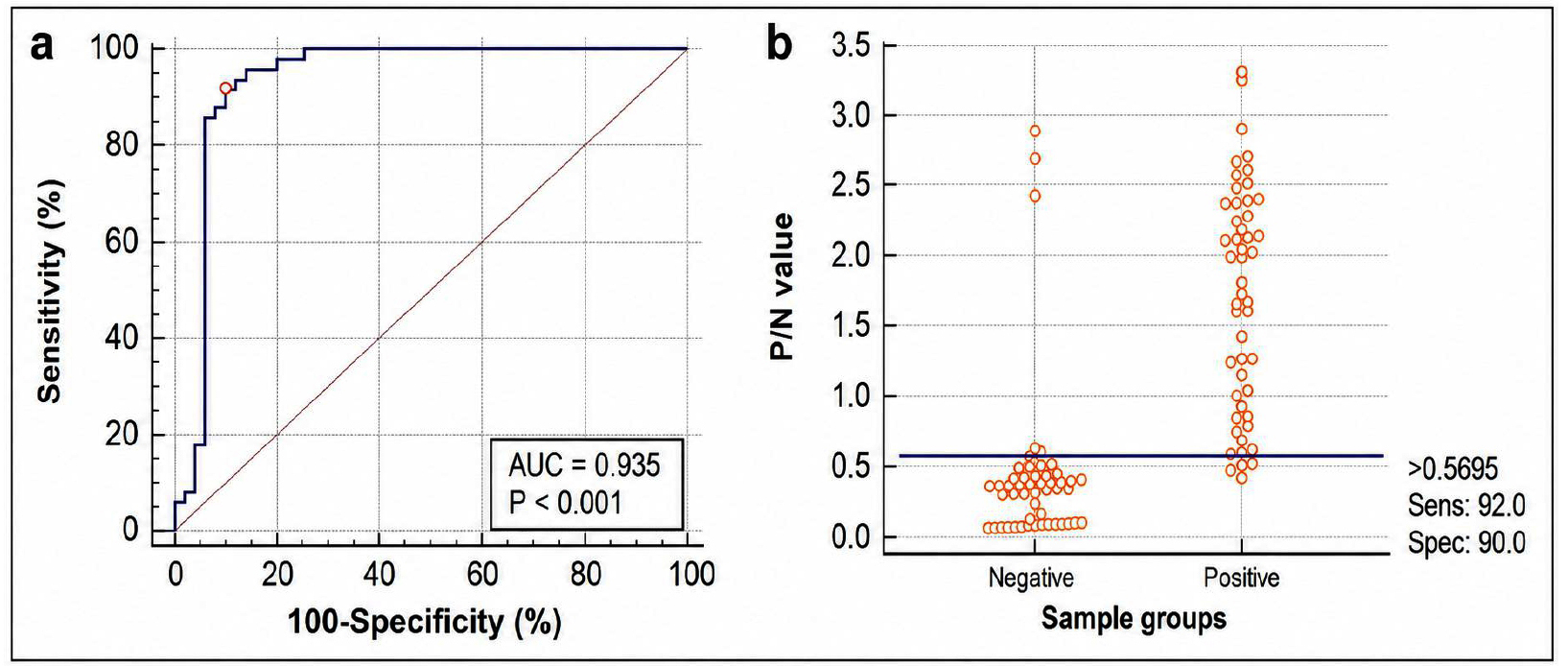
Figure 7. (a) Receiver Operating Characteristic (ROC) analysis of the developed rE2-iELISA against the Priocheck CSFV antibody detection kit. (b) Distribution of positive and negative serum samples based on P/N values with the selected cutoff point
Comparative analysis of the ROC curves indicated a statistically significant difference in diagnostic performance between the two assays, with an AUC difference of 0.216 (SE = 0.0482; 95% CI: 0.122-0.311). This difference was highly significant (z = 4.484; P < 0.0001), indicating a substantially higher diagnostic accuracy of the rE2 iELISA relative to the commercial Priocheck CSFV Antibody ELISA.
Accurate and prompt detection of Classical Swine Fever Virus (CSFV) in pig serum samples is pivotal for effective epidemiological surveillance, immune-prophylaxis assessment, and implementation of control strategies. The current research outlines a comprehensive strategy for the development of a rE2 iELISA for serological detection of CSFV antibodies in swine. The E2 glycoprotein is well documented as the main epitope for virus-neutralizing immunoglobulins and, consequently, has been broadly used in Sero-epidemiological diagnostics and immunogenicity evaluation frameworks.6,20 The BCAD region of the CSFV E2 glycoprotein was selected as both conformation-dependent and linear epitopes are predominantly present within the B/C/D/A domains in the N-terminal half of E2, which are responsible for generating neutralizing antibodies in infected pigs21,22 as this region is having considering its immunodominant properties. Conserved nature and reported diagnostic significance. The E2 gene has been uncovered to encode highly conserved immune targets within the BCAD domain in recent computational and evolutionary analysis, which indicates that it is a stable antigenic target for serodiagnosis applications. Pan-genomic entropy profiling of the E2 glycoprotein showed that the BCAD region has a comparatively reduced level of sequence variability and contains conserved epitopic regions that are crucial for antibody recognition.23 Further, an extensive phylogenetic and molecular evolutionary analysis of the CSFV E2 gene revealed that the BCAD region is highly conserved despite genetic diversifications among circulating genotypes, suggesting its use in the development of diagnostic assays.24,25 Previous research has shown that the E2 glycoprotein, notably the BCAD domain serves as a key immunogenic region capable of producing substantial humoral immune responses, making it an ideal choice for ELISA-based antibody detection methods.23,24 The present study commenced with the rational design of primers targeting the immunodominant BCAD region of the E2 glycoprotein, a domain recognized for its pronounced antigenicity and sequence conservation among diverse CSFV isolates. Using this rE2 glycoprotein confers substantial benefits relative to whole-virion or crude antigenic extracts, including enhanced Bio-safety profiles, inter-batch reproducibility, and markedly diminished serological cross-reactivity with heterologous pestiviruses such as bovine viral diarrhea virus (BVDV) and border disease virus (BDV). The successful amplification of the 546 bp target fragment and its subsequent cloning into the pET-28a expression vector confirmed the suitability of the selected region for recombinant antigen production.
Expression of the rE2 protein in E. coli BL21(DE3) yielded a protein of approximately 25 kDa, consistent with the predicted molecular weight of the BCAD domain fused with a His-tag. The protein was predominantly recovered in the insoluble fraction, a common occurrence for viral glycoprotein domains produced in prokaryotic hosts. Purification was achieved using Ni-NTA affinity chromatography following solubilization, resulting in efficient recovery of the recombinant protein. Crucially, the presence and antigenic integrity of the purified protein were validated by Western blot analysis, which confirms the specific reactivity with both anti-His antibodies and CSFV convalescent serum. These findings confirm the preservation of diagnostically relevant epitopes within the expressed recombinant antigen.
An indirect ELISA was developed and standardized using the purified rE2 protein as the coating antigen. Antigen concentration, serum and conjugate dilutions, blocking conditions and incubation timings were all systematically optimized to produce a repeatable assay with predictable signal characterization and low background reactivity. The development of reliable and repeatable ELISA systems has been revealed that it relies on parallel optimization techniques.26 The current study’s optimized parameters showed consistent OD readings with low intra and inter-assay coefficients of variation, indicating the high analytical precision of the assay. Previous investigations have confirmed that E2-based ELISAs provide better analytical sensitivity and diagnostic specificity than immunoassays using other structural and non-structural viral proteins.27,28 Concordant with these findings, the developed rE2 iELISA herein showed the clear discrimination between positive and negative samples with minimal nonspecific background signal. Receiver Operating Characteristic (ROC) curve analysis was employed to determine the diagnostic cut-off, which produced an objective and statistically valid threshold that minimized sample misclassification and ensured an optimal balance between sensitivity and specificity. In the validation of diagnostic platforms, the choice of ROC-derived cut-off values is widely supported since it ideally provides analytical sensitivity and specificity reducing both false-positive and false-negative classifications.29 A panel of 500 field serum samples was used to further evaluate the assay’s diagnostic performance, showing its suitability in field conditions. Comparative evaluation with the indigenously produced iELISA found nearly complete agreement, as evidenced by an AUC value of 0.995, as well as high sensitivity and specificity. That degree of agreement highlights the new assay’s reliability and supports its prospective use as a regional diagnostic alternative. The higher area under the curve (AUC) obtained in this study highlights that the proposed assay has good diagnostic fidelity, which concurs with the performance criteria of previously characterized CSFV E2- targeted ELISA techniques.30,31 Checkerboard titration confirmed particular antigen-antibody interaction by showing a dilution-dependent shift in OD values for positive and negative sera across all antigen concentrations. Despite lower dilutions resulted in high OD values and fold differences, these conditions were close to signal saturation and displayed enhanced background reactivity. At serum dilution of 1:100, OD values remained within the linear dynamic range, while negative sera showed modest background signals (<0.20). Fold difference showed accurate differentiation between positive and negative sera and the optimal signal-to-noise threshold was observed at a working dilution of 100 ng/well, with a fold difference of around 5.2. Subsequently, 100 ng/well antigen concentration blended with 1:100 serum dilution was selected as the optimal evaluation condition for further validation.
Evaluation of the developed rE2 iELISA with field-derived serum samples showed a high degree of concordance with established reference assays, thereby affirming its operational suitability in field diagnostic conditions. Consistent outcomes have been previously documented, wherein recombinant E2-based ELISAs have exhibited robust agreement with both commercially available kits and virus neutralization assays.32,33 When assessed alongside the commercial Priocheck CSFV Antibody ELISA kit, the rE2 iELISA also exhibited strong diagnostic accuracy; however, statistically significant differences in AUC values indicated that the commercial kit performed superiorly. These discrepancies may be attributable to differences in assay format, antigen presentation, and the extent of commercial optimization.
Despite these differences, the rE2 iELISA exhibited diagnostic performance within an excellent range and presents several practical advantages. By using a recombinant antigen, batch-to-batch consistency will be maintained, handling infectious materials minimized and biosafety regulation are simplified. The assay is also convenient and suitable for high-throughput testing, which makes it suggested for routine Sero-epidemiological surveillance, vaccine efficacy monitoring and through epidemiological investigations. It is especially valuable in scenarios with scarce resources. All of these findings show that the developed indigenous ELISA is a dependable and useful tool to support CSFV surveillance and control programs. Further interlaboratory validation and comparative evaluation across a range of CSFV genotypes will enhance its usefulness and improve its potential as a diagnostic application.
The developed rE2 iELISA is more suitable as a sensitive, specific, and reliable serological tool for large-scale CSF serosurveillance and control programs. Comparative ROC analysis against reference ELISA assays demonstrated that the developed rE2 iELISA exhibits robust diagnostic accuracy and sensitivity comparable to established commercial and indigenous assays. The recombinant antigen-based approach offers significant advantages including simplicity in production, cost-effectiveness, and potential for optimization and adaptation to resource-limited settings, thereby contributing to global efforts in minimizing the economic and epidemiological impact of Classical Swine Fever on the swine industry worldwide.
ACKNOWLEDGMENTS
The authors gratefully acknowledge the financial and institutional support provided by the Indian Council of Agricultural Research (ICAR), Department of Agricultural Research and Education (DARE), Government of India, and the Department of Animal Husbandry and Dairying. The authors also thank the Vice Chancellor, TDU, Bengaluru, for their valuable guidance and support throughout the study.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
MJ and SSP conceptualized the study. SSP applied methodology, collected resources, performed validation and supervised the work. MJ performed data curation, formal analysis and investigation. MJ wrote the original draft. MJ and SSP wrote the manuscript. PN, KPS, MJ, SSP and CBM reviewed and revised the manuscript. All authors read and approved the final manuscript for publication.
FUNDING
This study was supported by the Department of Animal Husbandry and Dairying (DAHD), Government of India, New Delhi, under the Livestock Health and Disease Control Programme (LHDCP), grant number K-11053(5313)/21/2019-LH (E-14082).
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
Not applicable.
- Moennig V. The control of classical swine fever in wild boar. Front Microbiol. 2015;6:1211.
Crossref - Postel A, Austermann-Busch S, Petrov A, Moennig V, Becher P. Epidemiology, diagnosis and control of classical swine fever: Recent developments and future challenges. Transbound Emerg Dis. 2018;65:248-261.
Crossref - Tautz N, Tews BA, Meyers G. The molecular biology of pestiviruses. Adv Virus Res. 2015;93:47–160.
Crossref - Malik YS, Bhat S, Kumar ORV, Yadav AK, Sircar S, Ansari MI, Sarma DK, Rajkhowa TK, Ghosh S, Dhama K. Classical Swine Fever Virus Biology, Clinicopathology, Diagnosis, Vaccines and a Meta-Analysis of Prevalence: A Review from the Indian Perspective. Pathogens. 2020;9(6):500.
Crossref - Sánchez O, Barrera M, Rodríguez MP, et al. Classical swine fever virus E2 glycoprotein antigen produced in adenovirally transduced PK-15 cells confers complete protection in pigs upon viral challenge. Vaccine. 2008;26(7):988-997.
Crossref - Paton DJ, McGoldrick A, Greiser-Wilke I, et al. Genetic typing of classical swine fever virus. Vet Microbiol. 2000;73(2-3):137-157.
Crossref - Luo Y, Li S, Sun Y, Qiu HJ. Classical swine fever in China: A minireview. Vet Microbiol. 2014;172(1-2):1-6.
Crossref - Wang L, Madera R, Li Y, McVey DS, Drolet BS, Shi J. Recent Advances in the Diagnosis of Classical Swine Fever and Future Perspectives. Pathogens. 2020;9(8):658.
Crossref - Yang Z, Li L, Pan Z. Development of multiple elisas for the detection of antibodies against classical swine fever virus in pig sera. Virol Sin. 2012;27(1):48-56.
Crossref - Yi W, Zhu H, Wu Y, et al. The recombinant Erns and truncated E2-based indirect enzyme-linked immunosorbent assays to distinguishably test specific antibodies against classical swine fever virus and bovine viral diarrhea virus. Virol J. 2022;19(1):121.
Crossref - Meyer D, Fritsche S, Luo Y, et al. The double-antigen ELISA concept for early detection of Erns-specific classical swine fever virus antibodies and application as an accompanying test for differentiation of infected from marker vaccinated animals. Transbound Emerg Dis. 2017;64(6):2013-2022.
Crossref - Greiser-Wilke I, Blome S, Moennig V. Diagnostic methods for detection of classical swine fever virus—status quo and new developments. Vaccine. 2007;25(30):5524-5530.
Crossref - Kaiser V, Nebel L, Schüpbach-Regula G, Zanoni RG, Schweizer M. Influence of border disease virus (BDV) on serological surveillance within the bovine virus diarrhea (BVD) eradication program in Switzerland. BMC Vet Res. 2017;13(1):25.
Crossref - Clavijo A, Lin M, Riva J, Mallory M, Lin F, Zhou EM. Development of a competitive ELISA using a truncated E2 recombinant protein as antigen for detection of antibodies to classical swine fever virus. Res Vet Sci. 2001;70(1):1-7.
Crossref - Chen N, Tong C, Li D, et al. Antigenic analysis of classical swine fever virus E2 glycoprotein using pig antibodies identifies residues contributing to antigenic variation of the vaccine C-strain and group 2 strains circulating in China. Virol J. 2010;7:378.
Crossref - Li W, Mao L, Yang L, Zhou B, Jiang J. Development and partial validation of a recombinant E2-based indirect ELISA for detection of specific IgM antibody responses against classical swine fever virus. J Virol Methods. 2013;191(1):63-68.
Crossref - Naik N, Hiremath J, Kumar HBC, et al. Prevalence of classical swine fever, African swine fever, and Japanese encephalitis: a multi-disease study in Indian pigs. Veterinaria México OA. 2025;12.
Crossref - Patil SS, Muruganantham V, Jogaiah M, et al. Post-vaccination seroconversion and lineage-level molecular characterization of classical swine fever virus in five northeastern states of India. Trop Anim Health Prod. 2026;58(2):167.
Crossref - Zweig MH, Campbell G. Receiver-operating characteristic (ROC) plots: A fundamental evaluation tool in clinical medicine. Clin Chem. 1993;39(4):561–577.
Crossref - Lin M, Lin F, Mallory M, Clavijo A. Deletions of structural glycoprotein E2 of classical swine fever virus strain alfort/187 resolve a linear epitope of monoclonal antibody WH303 and the minimal N-terminal domain essential for binding immunoglobulin G antibodies of a pig hyperimmune serum. J Virol. 2000;74(24):11619-11625.
Crossref - Chang CY, Huang CC, Deng MC, et al. Identification of conformational epitopes and antigen-specific residues at the D/A domains and the extramembrane C-terminal region of E2 glycoprotein of classical swine fever virus. Virus Res. 2012;168(1-2):56-63.
Crossref - Chang CY, Huang CC, Lin YJ, et al. Antigenic domains analysis of classical swine fever virus E2 glycoprotein by mutagenesis and conformation-dependent monoclonal antibodies. Virus Res. 2010;149(2):183-189.
Crossref - Ganesh KR, Patil SS, Harish J, Suresh KP, Hiremath J, Nayakvadi S. Pan-genomic entropy profiling reveals conserved immune hotspots in the E2 glycoprotein of Classical Swine Fever Virus. Veterinary Vaccine. 2026;5(1):100160.
Crossref - Patil SS, Harish J, Ramakrishna GK, Suresh KP, Hiremath J, Nayakvadi S. Comprehensive E2 gene-based phylogeny uncovers emerging lineages and refines global genotype framework of Classical Swine Fever Virus. Virology. 2026b;619:110875.
Crossref - Patil SS, Harish J, Ramakrishna GK, et al. Molecular evolution, codon usage bias, and population differentiation of the E2 gene in Classical Swine Fever Virus. J Adv Biol Biotechnol. 2026c;29(4):457-469.
Crossref - Crowther JR. The ELISA Guidebook. 2nd ed. Methods in Molecular Biology. Vol 516. Humana Press, New York; 2009.
Crossref - Van Rijn PA, van Gennip HGP, de Meijer EJ, Moormann RJM. Epitope mapping of envelope glycoprotein E2 of classical swine fever virus. J Gen Virol. 1996;77:2053-2061.
Crossref - Deng MC, Huang CC, Huang TS, et al. Phylogenetic analysis of classical swine fever virus isolated in Taiwan. Vet Microbiol. 2005;106(3-4):187-193.
Crossref - Greiner M, Pfeiffer D, Smith RD. Principles and practical application of the receiver-operating characteristic analysis for diagnostic tests. Prev Vet Med. 2000;45(1-2):23-41.
Crossref - Liu L, Xia H, Wahlberg N, Belak S, Baule C. Phylogeny, classification and evolutionary insights into pestiviruses. Virology. 2011;385(2):351-357.
Crossref - Blome S, Staubach C, Henke J, Carlson J, Beer M. Classical swine fever—An updated review. Viruses. 2017;9(4):86.
Crossref - Moser C, Ruggli N, Tratschin JD, Hofmann MA. Detection of antibodies against classical swine fever virus in swine sera by indirect ELISA using recombinant envelope glycoprotein E2. Vet Microbiol. 1996;51(1-2):41-53.
Crossref - Ji S, Luo Y, Zhang T, et al. An improved indirect ELISA for specific detection of antibodies against classical swine fever virus based on structurally designed E2 protein expressed in suspension mammalian cells. Arch Virol. 2018;163(7):1831-1839.
Crossref
© The Author(s) 2026. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.