


ISSN: 0973-7510
E-ISSN: 2581-690X
Diarrheagenic Escherichia coli, an important etiologic agent of diarrhea is a major public health problem in developing countries, particularly in children. Enteropathogenic Escherichia coli is a leading cause of infantile diarrhea. Although the frequency of these organisms has decreased, they continue to be an important cause of diarrhea. Therefore, in the present study, a sensitive and specific IgY mediated Immunocapture-PCR (IC-PCR) was developed for the detection of enteropathogenic E. coli (EPEC). Due to an advantage of avian immunoglobulin (IgY) to have the least affinity towards staphylococcal enterotoxin A (SpA) responsible for false positives, we employed anti- outer-membrane protein (OMP) IgY generated in chicken for capture of bfpA gene and was incorporated with PCR amplification. In the present study, IgY mediated immunocapture of bfpA gene was free from false positives due to protein A, a common drawback in IgG mediated immunocapture techniques. Furthermore, spiking studies and analysis on natural samples emphasized the robustness as well as applicability of developed method. The developed assay could be reliable in the detection of EPEC as a routine investigation method. Further, the assay could be further applied for the detection of other pathotypes from food and clinical sources.
EPEC, Immunoglobulin Y, Staphylococcal protein A, Immunocapture-PCR, E. coli, Diarrheagenic E. coli.
Diarrheal illness has been a major health concern worldwide, with 1.7 billion diarrheal cases occurred every year. It is also the second leading cause of death in children1. Of the diarrheagenic E. coli, EPEC is the first pathotype to be identified and is a major cause of diarrhea in children below 5 years of age and was epidemiologically related during outbreaks in the 1940s and 1950s2. EPEC remains a major cause of infantile diarrhea in low-income countries, accounting for 800,000 fatalities per year worldwide3. In developing countries, healthy carriage of enteric pathogen is attributed to many factors including host susceptibility (related to the child’s age, breastfeeding, nutritional and immunological status), bacterial virulence factors (different virulence genes) and environmental factors (poor hygiene, and high fecal contamination)4. Recent studies shows that a high prevalence (48.1%) of EPEC in Northern region of India. The prevalence of EPEC infection varies among different geographical areas and also based on differences in age distribution, study populations and also socio-economic class may also contribute to the epidemiology of infection5.
Unlike other pathotypes, EPEC is a non-invasive pathotype which does not produce enterotoxins and results in a characteristic ‘attaching and effacing’ (A/E) lesions on the surface of the host’s intestinal epithelium6. Previous studies have implicated the presence of EAF (EPEC adherence factor) plasmid in strains that were associated with outbreaks, which harbours an operon responsible for the biogenesis of type IV bundle forming pili (bfp). bfpA encodes for the major structural subunit of BFP, ‘bundlin’ and its expression is regulated at transcriptional level7.
Traditional diagnosis involving O:H serotypes has been extensively used for the detection of EPEC over the years. Based on phenotypic properties of EPEC, histopathological techniques targeting A/E using microscopy and cell culture techniques were used. Hep-2 cell adherence assay and FAS tests were originally used to detect EPEC, however, it cannot be routinely used because of the unavailability of tissue culture facilities in most of the laboratories2. In addition to this, genotypic tests by targeting major genes, i.e., eae, bfpA and stx have also been adopted due to their sensitivity and specificity. Molecular methods like PCR, DNA probe hybridization, RT-PCR techniques and immunoassays including ELISA, Western blot have also been routinely used. However, many of these methods are cost-ineffective and hence cannot be employed for regular testing and need technical expertise to carry out. On the other hand, immunocapture PCR (IC-PCR) technique has been well-established in diagnosis for detection of various bacterial, viral pathogens and also mycotoxins and disease specific antibodies (8, 9). In this method, antibodies along with nucleic acid amplification are used to detect proteins, in which gene amplification helps in signal enhancement. In addition to this, IC-PCR is more sensitive than quantitative PCR due to several washing steps prior to addition of DNA marker.
Outer membrane proteins (OMP) of Gram negative bacteria consists of approximately 50% of the total outer-membrane mass10. OMPs contain an integral membrane proteins and lipoproteins, which play a vital role in adhesion, invasion, bacteriophage receptors, resistance to antibiotics and maintaining the integrity11. They are also essential in bacterial pathogenesis because they increase the pathogen to adapt in host niches. OMPs so far described in EPEC are found to be virulence factors that include intimin and Efa1/LifA acting as adhesins12.
Considering the aforementioned factors, we developed a highly sensitive and specific assay by coupling ELISA with PCR, IC-PCR for the detection of bfpA of EPEC pathotype from culture supernatents and contaminated food samples using chicken IgY as capture antibo0dy in a 96-well PCR plate format. To achieve this, we targeted OMP for the production of polyclonal antibodies. Further, we selected bfpA gene for specific detection for the specific detection of EPEC. IC-PCR format was also evaluated on artificially spiked food and clinical stool samples to assess sensitivity and specificity in detection of EPEC.
Dehydrated bacteriological media, selective media and supplements were procured from HiMedia laboratories, Mumbai, India. Taq DNA polymerase and dNTPs were purchased from Sigma Aldrich, India. All the inorganic chemicals used in various buffers were of analytical grade and procured either from Merck, India or Sisco Research Laboratories Pvt. Ltd. India. Microtiter plates used for sample incubation and immunocapture step were from Nunc, USA. HRP-conjugated anti-chicken raised in rabbit was procured from Sigma Aldrich, India. Nitrocellulose membrane was from Pall Life Sciences, USA.
Outer membrane protein (OMP) extraction
Outer membrane proteins of E. coli ATCC 35401 strain was extracted using N-lauoryl sarcosine (Sarkosyl) according to the method described by Hobb et al., 2009 with minor modifications. Briefly, bacterial cells from 500 ml overnight culture was harvested and cells were washed in 1x PBS (pH-7.4) followed by centrifugation at 10,000 rpm for 5 min. Cell pellet was resuspended in 10 ml of 10 Mm Tris-Cl and ruptured by sonication. During sonication, the cells were incubated in ice and 1 mM PMSF was added to avoid protein denaturation. The ruptured membranes were collected by centrifugation at 17,000 rpm maintained at 4 °C for 1 h and resuspended in 3 ml of 1% Sarkosyl (HiMedia, India) in 10 mM Tris-Cl solution and incubated overnight at 4 °C. Following incubation, the protein was centrifuged at 17,000 rpm for 1 h at 4°C. Protein pellet was washed with 10 mM Tris followed by another round of centrifugation as described above. Finally, the protein pellet was resuspended in 1 ml of 10 mMTris-Cl, pH-7.4. Protein quantity was estimated by Bradford assay using bovine serum albumin as standard13.
Preparation of chicken antibodies (IgY) against outer-membrane protein (OMP) of EPEC
Four-week-old chickens were immunized with 200 µg of OMP preparation in emulsion with Freund’s complete adjuvant (FCA) (Sigma-Aldrich) and immunized via intramuscular injection. Same concentration in emulsion with Freund’s incomplete adjuvant was given in four booster doses in 15 days intervals. Antibody titre was determined ten days after last immunization by indirect ELISA in 96 well plates using OMP preparation as antigen. Eggs were collected and chicken antibodies were extracted from eggs using PEG precipitation method after fourth immunization14.
Reactivity testing by Western blot analysis
The pattern of reactivity of anti-OMP IgY antibodies with different bacterial species, including pathotypes of E. coli was evaluated by Western blot analysis. Bacterial species belonging to Enterobacteriaceae were grown overnight in BHI broth and cells were harvested by centrifugation. Whole cell lysate was prepared by resuspending the cell pellet in equal volumes of PBS and 2x Laemmli buffer. Bacterial suspension was subjected to heating for 10 min on a boiling water bath. Cell lysate was clarified by centrifugation at 10,000 RPM for 10 min and subjected to SDS-PAGE on a Cleaver Mini dual vertical gel electrophoresis unit. After separation, proteins were transferred onto nitrocellulose membrane using Cleaver blotting apparatus. Membranes were blocked with 5 % skim milk solution for 2 h at RT. After washing the milk solution in PBST (PBS with 0.05% Tween 20), the membrane was probed with 1:500 dilutions of anti-OMP IgY antibodies for 30 min at RT. Following wash with PBST, membrane treated with IgY was incubated with peroxidase-conjugated anti-chicken secondary antibodies raised in rabbit (Sigma). After washing in PBST solution, membrane was developed with DAB tetrahydrochloride and 0.03% H2O2 in PBS. The reaction was stopped by flooding the membrane in distilled water after 2-3 minutes in developing solution.
Reactivity testing by dot-ELISA
To assess the specificity of generated chicken antibodies, dot ELISA was performed with diarrheagenicE. coli pathotypes by using reference strains: ATCC 35401 (ETEC), ATCC 43887 (EPEC), ATCC 43893 (EIEC) and USFDA (EHEC). A clinical isolate (SC-542) which was confirmed by monoplex PCR for the presence of aggR gene was sequenced and employed as the reference strain for EAEC in the study. Briefly, the cultures were grown in BHI broth overnight and cells from 1 ml broth were harvested by centrifugation. Cells were resuspended in 100 µl of 50 mM carbonate buffer (pH-9.8), 10 µl of each strain was spotted onto nitrocellulose membrane followed by blocking in 5 % skim milk solution for 1 h at room temperature. The membranes were then treated with 1:500 dilutions of anti-OMP chicken antibodies in PBS for 30 min followed by peroxidase labelled anti- chicken rabbit secondary antibodies. Finally, the blots were developed similar to Western blot as mentioned in above.
Sandwich ELISA
Sandwich ELISA test was performed in 96-well microtiter plates (Nunc). Plates were coated with1:500 dilution of IgY (5 mg/mL) in 0.05 M bicarbonate buffer (pH 9.8) as capturing antibody, incubated overnight at 4 ºC and blocked with 5% non-fat skim milk for 1 h. The plates were washed with PBST. Different dilutions of EPEC cells (109-100) were added to the wells followed by incubation at 37 ºC for an hour. Sterile PBS was used as blank. 1 ml of anti-OMP mice antibody (in-house generated) was added as revealing antibody and incubated for an hour at 37 ºC. Plates were washed in PBST. Peroxidase-conjugated anti-mouse goat antibody was used as secondary antibodies. The plates were then washed thoroughly and finally developed with chromogenic substrate o-phenylenediaminedihydrochloride (OPD) (Sigma) (0.5 mg/mL) and 0.03% H2O2 in 0.2M citrate-phosphate buffer (pH 5.0). The reaction was stopped by 2M H2SO4 and the absorbance was measured at 450 nm using Infinite M200 Pro multimode microtiter plate reader (Tecan).
PCR using EPEC-specific primers
Genomic DNA samples extracted by boil-lysate technique from reference strain of EPEC was used for PCR. The in-house primers targeting bfpA gene was employed in the study (Data not published). The optimized protocol was carried out with a 20 µL mixture containing 1X PCR buffer, 1.5 mmol-1 MgCl2, 0.20 mmol-1 dNTPs, 1.25 U of Taq DNA polymerase, 5 pmol of each primer and 100 ng of each of the template DNA samples. The PCR conditions for amplification are as follows: initial denaturation at 94°C for 4 min, 30 cycles of denaturation at 94°C for 1 min, 53°C of annealing for 1 min, 72°C of extension for 1 min followed by final extension at 72°C for 8 min. Amplification of PCR products was visualized by gel electrophoresis using 2% (wt/vol) agarose gel (Seakem LE) in 0.5x Tris-borate EDTA buffer with 0.5 µg/mL ethidium bromide at 100 V for 45 min. The primers were designed using GeneRunner 5.1 by choosing specific regions with the help of BLAST software (Table 1).
Table (1):
Primer sequence for the detection of EPEC.
Gene |
Primer sequence (5′-3′) |
Amplicon size (bp) |
|---|---|---|
bfpA |
F-GCAGGTGTGATGTTTTACTA R- CAGCAGGAGTAATAGCAGTC |
430 |
Standardization of IC-PCR
IC-PCR was standardized with anti-OMP IgY antibodies, generated using purified OMP fraction from EPEC ATCC 35401 strain. Anti-OMP chicken antibodies were diluted at 1:500 dilutions in 50 mM carbonate-bicarbonate buffer. 96 well PCR tubes were coated with 20 µl of diluted IgY antibodies and allowed to coat at 4 °C overnight. The strips were washed in PBST and blocked with 5% skim milk for 1 h at 37 °C. Strips were washed in PBST to remove excess milk, air dried and stored at -20 °C for further use. During immunocapture, 20 µl of the sample (culture supernatants/enriched samples/extracted samples) was added to the antibody coated PCR plates and incubated for 30 min at RT followed by gentle washing in PBST 2-3 times and a final wash in distilled H2O. 20 µl of PCR master mix consisting of 1 x Taq buffer, 1.5 mM MgCl2, 100 µM, dNTPs, 5 pmol each of forward and reverse primers of bfpA gene and 1 U of Taq DNA polymerase was added to the PCR strips and subjected to PCR. The cycle conditions were as follows: initial denaturation at 94 °C for 5 min followed by 30 cycles of denaturation at 94 °C for 1 min, annealing at 53 °C for 1 min, extension at 72 °C for 30 sec and a final extension at 72 °C for 5 min. The PCR products were resolved on 1.5% agarose gel (Seakem LE) in 0.5x Tris-borate EDTA buffer containing 0.5 µg/ml ethidium bromide at 100 V for 45 min. The amplicons were examined and documented using GelDoc system (Syngene, UK).
Sensitivity and specificity testing
The specificity of the IC-PCR assay was determined by testing other closely related members of Enterobacteriacae as well as other Gram negative and Gram positive organisms with EPEC specific primers. Sensitivity of the IC-PCR assay was evaluated by subjecting the overnight grown culture to ten-fold serial dilution and evaluating 20 µl of each dilution by IC-PCR as described above. Least dilution at which the PCR gave a positive result was considered the limit of detection of assay. The CFU count in each dilution was determined by using plate count agar (PCA).
Artificial spiking studies
Artificial spiking studies were undertaken to evaluate the IC-PCR assay for its possible application in direct evaluation of food or clinical samples for testing the presence of EPEC pathotype. Briefly, different food and clinical samples were collected and subjected to appropriate dilutions. Solid samples were macerated at 1g quantity in 10 ml saline and then tested for spiking studies. The diluted samples was spiked with EPEC and allowed to incubate for 30 min. Samples were centrifuged briefly at 1000 RPM for 1 min, 20 µl of the sample was added to antibody coated PCR tubes for capture. Further testing was carried out as described earlier.
Evaluation of IC-PCR on clinical isolates
IC-PCR was evaluated on a panel of different E. coli standard and isolated strains recovered from clinical stool samples from SDM College of Medical Sciences and Hospital, Dharwad city in Karnataka state, India (unpublished results). Overnight grown cultures in BHI broth were used for testing with IC-PCR. The IC-PCR results were cross verified with direct PCR evaluation with isolated genomic DNA from each strain. The results were interpreted qualitatively based on the presence or absence of PCR amplification post IC-PCR testing.
Diarrhea is a serious public health problem and a major cause of morbidity and mortality in infants and young children. Diarrheal diseases are most encountered in low and middle-income countries in Africa, Asia and Latin America with lethal outcomes mainly due to poor living conditions such as inadequate water supply, poor sanitation and environmental hygiene (4). Diarrheagenic E. coli strains are one of the most important etiological agents, evolved by the acquisition, through horizontal gene transfer, of a particular set of characteristics that have successfully persisted in the host (15).
Among E. coli pathotypes of DEC, EPEC is the first pathotype to be discovered and the term was used to describe a certain group of E. coli strains epidemiologically related to outbreaks of infantile diarrhea in the 1940s and 1950s (16). Although EPEC was a major cause of infantile diarrhea in developed countries, in the recent years, it is more prevalent in developing countries (2). Like other pathotypes, the prevalence of EPEC infection varies between epidemiological studies based on different geographic region and socioeconomic class. In addition, differences in study populations, age distributions and methods used for detection and diagnosis may also contribute to the epidemiology of EPEC induced diarrheal disease (3).
Several techniques like PCRs, ELISA, Hep2 assay, Western blotting, RT-PCR, quantitative IPCR have been reported for detection of EPEC in food and clinical samples. Although molecular methods like PCR are rapid and sensitive in the detection of DEC, they only rely on the amplification of a target sequence encoding the protein using thermostable DNA polymerase. In contrast, immunological methods such as Western blot technique and ELISAs detect the actual protein and are highly specific but not as sensitive as PCR.
The majority of the antibody based techniques employ mammalian monoclonal antibodies which are known to be associated with SpA leading to false positive results. Previous studies have employed avian IgY antibodies as capture antibody which have less affinity towards SpA to avoid false positive result in immunoassays (17, 18). In addition, avian immunoglobulins have proved to be effective in numerous immunological applications since large quantities of polyclonal antibodies can be generated with a low-cost (19, 20, 21). Therefore, in the present study, we have developed IC-PCR combining the versatility of ELISA with the sensitivity of nucleic acid amplification of PCR without inference of SpA protein.
Previous studies have revealed the presence of more than twenty diverse outer membrane proteins in E. coli, with different characteristics, including adhesion, invasion, bacteriophage receptors, resistance to antibiotics and permeability controls, as well as structural functions. Since OMPs are present on all the E. coli pathotypes, in our study, we have extracted OMP from a standard strain of EPEC for the subsequent detection of EPEC. The resultant outer membrane protein preparation of E. coli ATCC-35401 carried out by sarkosyl treatment was quantified by Bradford assay with BSA as standard. The amount of OMP extracted was quantified to be 10 mg/l of overnight grown culture. The concentration of crude OMP preparation was adjusted to 2.0 mg/ml in PBS followed by storing at -20°C for further experiments. Crude OMP preparation was subjected to SDS-PAGE analysis for analyzing the number of proteins recovered. After staining, a total of 6-7 bands were visualized in the PAGE gels.
Chickens were immunized with OMP along with Freund’s complete and incomplete adjuvants. The concentration of anti-OMP IgY was determined by indirect ELISA, IgY was found in blood of immunized chickens after the fourth immunization with a titer of 1:64,000 was observed using OMP as the antigen. Egg extraction was carried out after the fourth immunization. The extracted IgY by PEG precipitation method yielded more than 20 mg/ egg yolk and the concentration was adjusted to 5 mg/ml in PBS and stored at
-20 °C for further use. The IgY antibodies acts as a common epitope for all the pathotypes is mono-specific for OMP which facilitating the detection of DEC.
To check the efficacy of IgY and also cross-reactivity of OMP, Western Blot analysis was performed with all the major Enterobacteriaceae members by using chicken anti-OMP antibodies. The major pathotypes of E. coli tested for cross-reactivity reacted at 48 KDa along with other gram negative rods such as members of Salmonella and Shigella, Klebsiella pneumoniae, Enterobacter cloacea, Proteus mirabilis and Proteus vulgaris (Fig 1). Furthermore, reactivity of anti-OMP antibodies were evaluated by dot-ELISA with all the major Enterobacteriaceae members as presented in table 2. The majority of all the tested Gram negative rods (GNR) were reactive with IgY (Fig 2). Reactivity observed with majority of GNRs indicated conserved nature of outer membrane antigens due to their common ancestry. Also, there was no cross-reactivity with Gram positive members such as Staphylococcus, Listeria, and certain members of Bacillus species.
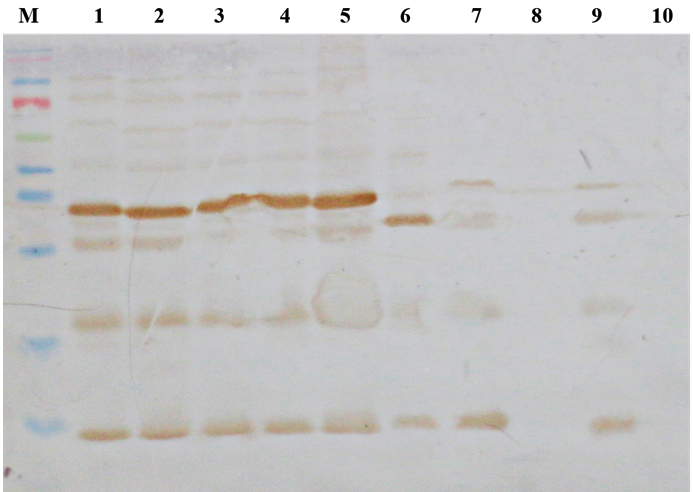
Fig. 1. Reactivity testing of anti-OMP-IgY by Western blot analysis. M — pre-stained protein ladder, L1-ETEC, L2— EHEC, L3—EPEC, L4—EAEC, L4—EPEC, L5— Salmonella typhimurium, L6 — Listeria monocytogenes, L7— Proteus vulgaris, L8— Klebsiella pneumonia

Fig. 2. Reactivity testing of anti-OMP-IgY by dot-ELISA. C-Control, 1- EIEC, 2- EAEC, 3-ETEC, 4-EPEC, 5- EAEC, 6- Salmonella typhimurium, 7- Klebsiella pneumonia, 8- Listeria monocyto-genes, 9- Staphylococcus aureus
Table (2):
List of various bacterial strains employed to determine specificity of IgY by dot-ELISA.
Bacterial Strain |
Result |
|
|---|---|---|
EPEC |
*USFDA-303 |
+ |
EHEC |
*ATCC 43893 |
+ |
EIEC |
*ATCC 43887 |
+ |
ETEC |
*ATCC 35401 |
+ |
EAEC |
*DFRL- SC-542 |
+ |
Bacillus subtilis |
* ATCC-6633 |
– |
Salmonella typhimurium |
DRDE-GWALIOR |
– |
Shigella spp. |
SDM-DHARWAD |
– |
Enterobacter cloacae |
NCIM 2015 |
– |
Klebsiella pneumonia |
ATCC 13883 |
– |
Listeria monocytogenes |
ATCC 19114 |
– |
Proteus vulgaris |
ATCC 33420 |
– |
Yersinia enterocolitica |
ATCC 23715 |
– |
Schematic representation of IC-PCR developed in the study is depicted in fig. 3. The described IC-PCR is based on capture of OMP by a surface bound IgY antibody followed by addition of whole cell lysate and pathotype-specific-primers (bfpA), of which these serve as a substrate in subsequent PCR step for signal generation. Initially, IC-PCR was standardized with culture supernatants of bfpA producing EPEC. After selecting the optimum annealing temperature of 53oC for the effective amplification of target genes, a master mixture consisting of primer pairs and reference genomic DNA of EPEC was added to each PCR reaction for detection of EPEC. A clear distinction of targeted gene amplicon was observed on 1.5% agarose gel electrophoresis (Fig. 4). The entire steps were performed with a minimum volume of 20 µl including coating of capture antibody, capture of antigen followed by PCR amplification. Therefore, the presence of few DNA marker molecules adhering to plates is sufficient in the PCR amplification. In addition, the error rate of IC-PCR is almost negligible than other PCR formats due to the several washing steps which facilitates the elimination of contamination.
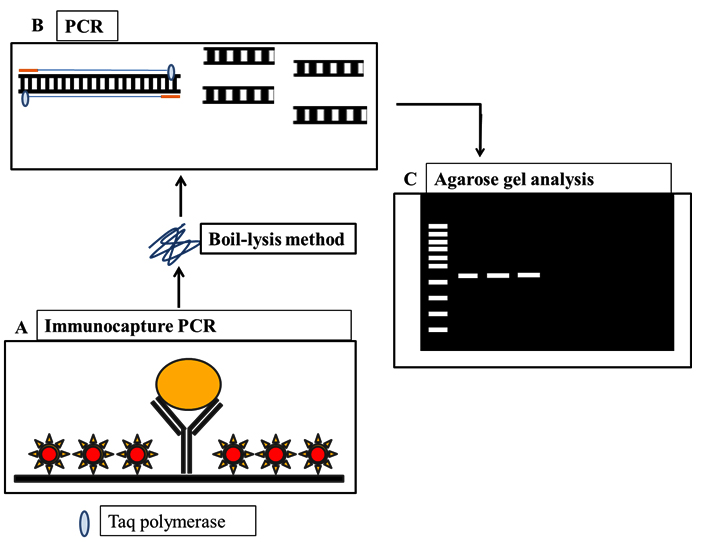
Fig. 3. Schematic representation of IC-PCR and PCR-ELISA employed in the present study.
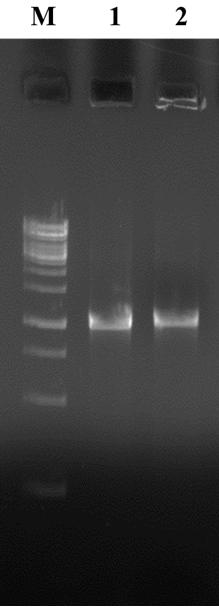
Fig. 4. Agarose gel electrophoresis. M- 100bp DNA Ladder , L1 and L2 – EPEC.
Table (3):
List of various bacterial strains employed to determine specificity by IC-PCR assay.
Bacterial Strain |
Source |
bfpA |
|---|---|---|
*USFDA-303 |
– |
|
*ATCC 43893 |
– |
|
Escherichia coli |
*ATCC 43887 |
+ |
*ATCC 35401 |
– |
|
*DFRL- SC-542 |
– |
|
Salmonella typhimurium |
DRDE-GWALIOR |
– |
Shigellaspp. |
SDM-DHARWAD |
– |
Enterobacter cloacae |
NCIM 2015 |
– |
Klebsiella pneumonia |
ATCC 13883 |
– |
Listeria monocytogenes |
ATCC 19114 |
– |
Proteus vulgaris |
ATCC 33420 |
– |
Yersinia enterocolitica |
ATCC 23715 |
– |
*E. coli reference strains
USFDA- U.S Food and Drug Administration, USA
ATCC – American Type Culture Collection, Rockville, MD, USA
DRDE-Gwalior- Defence Research and Development Establishment, Gwalior, India
SDM-DHARWAD – SDM College of Medical Sciences and Hospital, Dharwad, India
To increase specificity and sensitivity of IC-PCR, we have used IgY antibodies raised against OMP of E. coli and coupled with ELISA with PCR for an effective amplification of target product. Furthermore, our data revealed the assay to be highly specific with no cross reactivity with any other pathotype other than EPEC and also with other bacterial strains (Table 3). Further, E. coli specific OMP checked for cross reactivity with other enterobacteriacea members using pathotype-specific-primers of bfpA. Evaluation studies revealed that the primers designed for targeted gene of EPEC were highly specific and the results were reproducible. The sensitivity of IC-PCR performed by serially diluting bacterial cultures in 0.9 % saline solution ranging from 109 to 101 CFU/ml. The bacterial cell count enumerated on PCA. Further, the samples were examined by IC-PCR. The assay could detect cells at 102 CFU/ml proving it to be highly sensitive in detection and differentiation of EPEC (Fig. 5). The sensitivity achieved by this method was found to be far more than other methods. The method is also free from hindrances such as complex chemical composition of food matrix and inhibitors present in samples due to the presence of several washing steps. Further, the IC-PCR was tested on clinical strains of E. coli, the assay correlated 100% with PCR and ELISA. During artificial spiking studies, IC-PCR was able to detect bfpA from spiked food samples as low as 103 g/ml. The assay could successfully detect bfpA of EPEC from food even if the bacterial load is less than detectable amount, suggesting the potential application (Table 4).
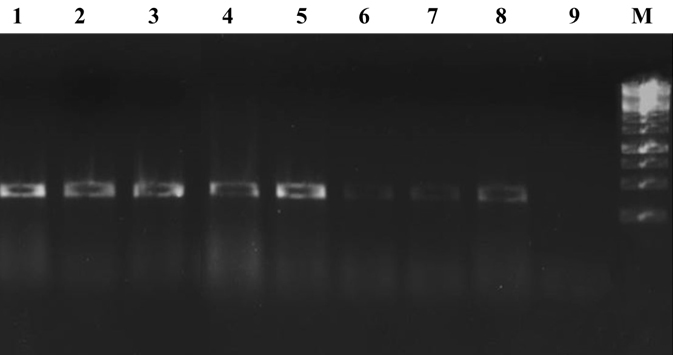
Fig. 5. Agarose gel electrophoresis showing sensi-tivity of IC-PCR. Lane 1 to 9- Sensitivity from 109 -101, M- 1kb DNA Lad-der.
Table (4):
List of naturally contaminated samples evaluated in the study.
Type of sample |
Number of samples tested |
Number of positive |
|---|---|---|
Milk |
30 |
9 |
Chicken |
13 |
3 |
Mutton |
09 |
1 |
Total |
52 |
13 |
The IC-PCR assay was compared with sandwich ELISA and PCR to assess its sensitivity. High sensitivity was obtained in IC-PCR compared to other assays. Immunocapture has enhanced the sensitivity and specificity whereas the specific primer pair used in PCR has intensified the signal. Therefore, the combined assay was better in terms of sensitivity and specificity compared to other two assays tested (Table 5). The evaluation of IC-PCR on clinical isolates revealed the presence of bfpA genes (n=4) among 70 DEC clinical isolates. However, the credibility of the assay was checked by confirming the results with a direct PCR and results matched with IC-PCR assay, when amplification was observed on 1.5% agarose gel electrophoresis.
Table (5):
Comparison of PCR, ELISA and IC-PCR.
Parameter |
Sandwich ELISA |
PCR |
IC-PCR |
|---|---|---|---|
Sensitivity |
105 |
103 |
102 |
The present method proved to be efficient with major improvements, the elimination of cross reactivity by employing IgY antibodies having less affinity towards SpA protein combined with high sensitivity. IgY antibodies from chickens are better alternatives to mammalian antibodies, due to their low cost production and the use of less invasive methods for their extraction and purification. The combinatorial approach of both molecular and immunological techniques revealed to be more efficient with higher sensitivity and specificity. The developed IC-PCR revealed to be efficient in detection of EPEC, on the other hand, the method can also be employed for detection of multiple analytes in a single PCR plate by differential capture antibody coating. The present study thus might provide a simple, sensitive and specific detection tool for detection of EPEC. The same strategy could be extended to detect other pathotypes of DEC and also other organisms.
ACKNOWLEDGMENTS
Authors are grateful to DRDO for providing the necessary facilities to carry out this work.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
- Sarker AR, Sultana M, Mahumud RA, Sheikh N, Van Der Meer R, Morton A. Prevalence and health care–seeking behavior for childhood diarrheal disease in Bangladesh. Global pediatric health. 2016; 3:2333794X16680901.
- Croxen MA, Law RJ, Scholz R, Keeney KM, Wlodarska M, Finlay BB. Recent advances in understanding enteric pathogenic Escherichia coli. Clinical microbiology reviews. 2013; 26(4):822-80.
- Hu J, Torres AG. Enteropathogenic Escherichia coli: foe or innocent bystander?. Clinical Microbiology and Infection. 2015; 21(8):729-34.
- Gomes TA, Elias WP, Scaletsky IC, Guth BE, Rodrigues JF, Piazza RM, Ferreira L, Martinez MB. Diarrheagenic Escherichia coli. brazilian journal of microbiology. 2016; 47:3-0.Ochoa TJ, Contreras CA.
- Enteropathogenic E. coli (EPEC) infection in children. Current opinion in infectious diseases. 2011; 24(5):478.
- Clements A, Young JC, Constantinou N, Frankel G. Infection strategies of enteric pathogenic Escherichia coli. Gut microbes. 2012; 3(2):71-87.
- Blank TE, Zhong H, Bell AL, Whittam TS, Donnenberg MS. Molecular variation among type IV pilin (bfpA) genes from diverse enteropathogenic Escherichia coli strains. Infection and immunity. 2000; 68(12):7028-38.
- Mehta PK, Raj A, Singh NP, Khuller GK. Detection of potential microbial antigens by immuno-PCR (PCR-amplified immunoassay). Journal of medical microbiology. 2014; 63(5):627-41.
- Waller DF, Ogata SA. Quantitative immunocapture PCR assay for detection of Campylobacter jejuni in foods. Applied and environmental microbiology. 2000; 66(9):4115-8.
- Niemeyer CM, Adler M, Wacker R. Immuno-PCR: high sensitivity detection of proteins by nucleic acid amplification. Trends in biotechnology. 2005; 23(4):208-16.
- Koebnik R, Locher KP, Van Gelder P. Structure and function of bacterial outer membrane proteins: barrels in a nutshell. Molecular microbiology. 2000; 37(2):239-53.
- Taddei CR, Oliveira FF, Piazza RM, Leme AF, Klitzke CF, Serrano SM, Martinez MB, Elias WP, Sant OA. Suppl 1: A Comparative Study of the Outer Membrane Proteome from an Atypical and a Typical Enteropathogenic Escherichia coli. The open microbiology journal. 2011; 5:83.
- Brasford MM. A rapid and sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem.. 1976; 72: 248-54.
- Pauly D, Chacana PA, Calzado EG, Brembs B, Schade R. IgY technology: extraction of chicken antibodies from egg yolk by polyethylene glycol (PEG) precipitation. Journal of visualized experiments: JoVE. 2011(51).
- Pavankumar AR, Sankaran K. The need and new tools for surveillance of Escherichia coli pathogens. Food Technology and Biotechnology. 2008 ; 46(2):125-45.
- Donnenberg MS, Whittam TS. Pathogenesis and evolution of virulence in enteropathogenic and enterohemorrhagic Escherichia coli. The Journal of clinical investigation. 2001; 107(5):539-48.
- Mizutani N, Sugita Konishi Y, Omoe K, Shinagawa K, Kawakami H, Kanno S, Sugiyama KI, Kamata Y. Advantages of immunoglobulin Y for the detection of Staphylococcal enterotoxin A in a double antibody sandwich enzyme linked immunosorbent assay. International journal of food science & technology. 2012; 47(1):155-9.
- Reddy PK, Shekar A, Kingston JJ, Sripathy MH, Batra H. Evaluation of IgY capture ELISA for sensitive detection of Alpha hemolysin of Staphylococcus aureus without staphylococcal protein A interference. Journal of immunological methods. 2013; 391(1-2):31-8.
- Tan NC, Tran H, Roscioli E, Wormald PJ, Vreugde S. Prevention of false positive binding during immunofluorescence of Staphylococcus aureus infected tissue biopsies. Journal of immunological methods. 2012; 384(1-2):111-7.
- Tan SH, Mohamedali A, Kapur A, Lukjanenko L, Baker MS. A novel, cost-effective and efficient chicken egg IgY purification procedure. Journal of immunological methods. 2012; 380(1-2):73-6.
- Reddy P, Ramlal S, Sripathy MH, Batra HV. Development and evaluation of IgY ImmunoCapture PCR ELISA for detection of Staphylococcus aureus enterotoxin A devoid of protein A interference. Journal of immunological methods. 2014; 408:114-22.
© The Author(s) 2018. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.