


ISSN: 0973-7510
E-ISSN: 2581-690X
Deepika Pandey Tiwari1,2, Poonam Mishra Chatterjee1,2, Niyati Uppadhyaya1![]() , Anirban Bhaduri1
, Anirban Bhaduri1![]() , Ritu Raval2
, Ritu Raval2![]() and Ashok Kumar Dubey1,2
and Ashok Kumar Dubey1,2 ![]()
https://dx.doi.org/10.22207/JPAM.13.2.03 | © The Author(s). 2019
Poly-γ-glutamic acid (PGA) is a homopolyamide, biosynthesized mostly by Bacillus sp. Our study focuses on understanding the genetic differences between the two species of Bacillus for their capability to produce PGA. Genes related to PGA synthesis, regulation, degradation and mannitol utilization of Bacillus subtilis Natto3 (BSN3) were compared with that of B. methylotrophicus IC4 (BMIC4). These strains differed in their genome sizes and average gene lengths. BMIC4 genome size was 4,214,684 bp which was larger than BSN3 comprising of 3,601,055 bp with no plasmid found in either of them. The average gene length of BSN3 and BMIC4 were 843.33 bp and 819.82 bp, respectively with higher number of predicted genes and proteins in BMIC4 (4341 and 4223 respectively). Interestingly, BMIC4 being larger in genome size and gene number, exhibited lesser number of unique pfam results (62) compared to 389 unique pfam of BSN3. Based on 16S rRNA gene sequence, BSN3 and BMIC4 were placed distantly on the phylogenetic tree. Sequence similarity of PGA producing genes ywsC, ywtA and ywtB between BSN3 and BMIC4 was 100%, 100% and 30% respectively. We report the presence of PGA degrading gene pgdS in BMIC4 which is otherwise reportedly absent in various strains of B. methylotrophicus. Sequence variation in the genes may have an impact on the PGA chain length, produced by these strains as BMIC4 produces high molecular weight PGA than BSN3. As B. methylotrophicus is newly discovered species, our comparative study will provide insights on the genomic variability between these two novel PGA producing strains.
Whole genome sequencing, Bacillus subtilis, Bacillus methylotrophicus, PGA, pgdS
Biopolymers have gained a lot of importance in the recent years due to their multifarious applications and biodegradability. Prospects of polyamino acids are being constantly considered to offer a great set of opportunities in various industrial sectors. Therefore, persistent efforts are being attempted in improving the microbial production of these biopolymers. PGA is one such biodegradable polymer that is produced in the g-form naturally by the Bacillus sp. due to its higher capacity for PGA production. PGA has wide range of industrial application such as in food, cosmetics, medical, wastewater treatment and protein crystallization1. Bacillus spp. are well known PGA producers but limited number of strains have been exploited at industrial scale out of which B. subtilis is one of the most utilized strains amongst all. One of the newly reported strains with potential to produce higher molecular weight PGA is B. methylotrophicus. The demand for ultra-high molecular weight PGA is slowly increasing in certain applications such as corneal wound healing, intenstinal Calcium absorption etc2-3. It is now being explored for PGA production due to the exigencies in the areas where higher molecular weight PGA is required.
B. subtilis is well-studied for the production and regulation of PGA4-7 but studies providing insights on PGA production using B. methylotrophicus are mostly recent and quite few. Genome of B. subtilis is substantially mapped 8 but such detailed studies on B. methylotrophicus are a few9-12. This triggered us to study and compare the genetic make-up of one of the most studied bacteria i.e. B. subtilis with the least explored microbe i.e. B.methylotrophicus.
B. subtilis and B. methylotrophicus are often associated with promoting the plant-growth13-14. B. methylotrophicus has been isolated from diverse sources like agricultural soil13, 15, wood tissues9 and also thrives in volcanic soils 16. Similarly, B. subtilis is omnipresent and has been isolated from various sources such as plant rhizosphere, soil, water ecosystems, food and animal colon17-18. We had isolated BSN3 from food and BCIM4 from soil. Both the bacteria then were studied for their nutritional preferences and genetic differences.
Nutrients for PGA production
Various Carbon and Nitrogen sources have been studied and optimized for Bacillus spp. such as B. subtilis, B. licheniformis, B. velezensis and B. methylotrophicus etc. where the reports reveal the use of D- and L-glutamic acid, sodium glutamate, glucose, glycerol, citric acid, maltose, sucrose, glutamine and a-ketoglutaric acid11-12,19-20. In one of our previous reports, we have mentioned the use of a rhizobacteria isolated from the agricultural field and identified as BMIC4 for PGA production. The fermentation media was supplemented with mannitol and monosodium glutamate along with peptone as nitrogenous source. Growth of BMIC4 and PGA production was observed to be supported by mannitol instead of simple sugars such as glucose, lactose, and maltose. BMIC4 produced 36 g/L PGA in the above mentioned media composition with mannitol as the main carbon source15.
PGA production by B. subtilis has been reported to use glutamate as substrate and glucose and citrate as carbon sources. Studies have also suggested that in certain conditions B. subtilis prefer citrate for PGA production over glucose12,21. Studies in other Bacillus spp. like B. licheniformis NCIM 2324, have also shown PGA production in a combination of citric acid, glycerol, glutamate and ammonium sulphate. Production of PGA was increased from 5.27 mg/ml (basal medium) to 26.1 mg/ml (test medium). In a media with glucose as main carbon source, B. licheniformis A13 produced 28.2 mg/ml PGA1. PGA production has also been shown to improve from 5.7 to 20.5 mg/ml when glycerol was added to the fermentation medium of Bacillus licheniformis ATCC 9945a22. Hence, great diversity in nutrient requirement exists between B. subtilis and B. methylotrophicus with respect to PGA production. To understand the variable PGA production due to different media compositions, comparative studies can play a crucial role.
Genetic studies
In Bacillus spp., a membrane bound PGA synthetase assembly is responsible for PGA production. This complex is coded by pgs operon containing ywsC and ywtABC genes (also known as pgsBCAE)1,4-5. We recently studied the expression dynamics of these genes in BSN3 in a media supplemented with the combination of glucose and glutamic acid and separately with glutamic acid alone. In the given conditions pgs operon showed a unique expression trend where, ywsC and ywtA genes were up-regulated and ywtB and ywtC were down-regulated7. This was a unique expression because in spite of being the part of the same operon selective up-regulation and down-regulation of the above mentioned genes was observed to take place. Therefore the presence or absence of specific substrate in the media could cause a direct or indirect impact on the up-regulation and down-regulation of PGA producing genes.
These novel outcomes and the dearth of knowledge, specifically in the case B. methylo-trophicus encouraged us to dig deeper insights at the molecular level and comparatively investigate the genes responsible for PGA production, regulation, degradation and mannitol utilization between BSN3 and BMIC4. To achieve this comparative analysis, we have carried out the whole genome sequencing of both our previously studied novel strains; BSN3 and BMIC4. A comparative analysis was thus carried out between the two above mentioned species to understand the variation of gene sequences, corresponding differences in the putative proteome which may ultimately affect various aspects of PGA production.
Strains, media and genomic DNA isolation
BMIC4 (NCIM 5536) isolated from the rhizosphere soil and BSN3 (NCIM 5537) isolated from a Japanese dish15 were propagated in 5 ml sterile Nutrient broth at 37°C overnight. Genomic DNA was isolated using (Fig. 1) DNeasy Blood & Tissue Kits (Qiagen) followed by RNAse A (Thermo Fisher Scientific) treatment as per manufacture’s protocol.
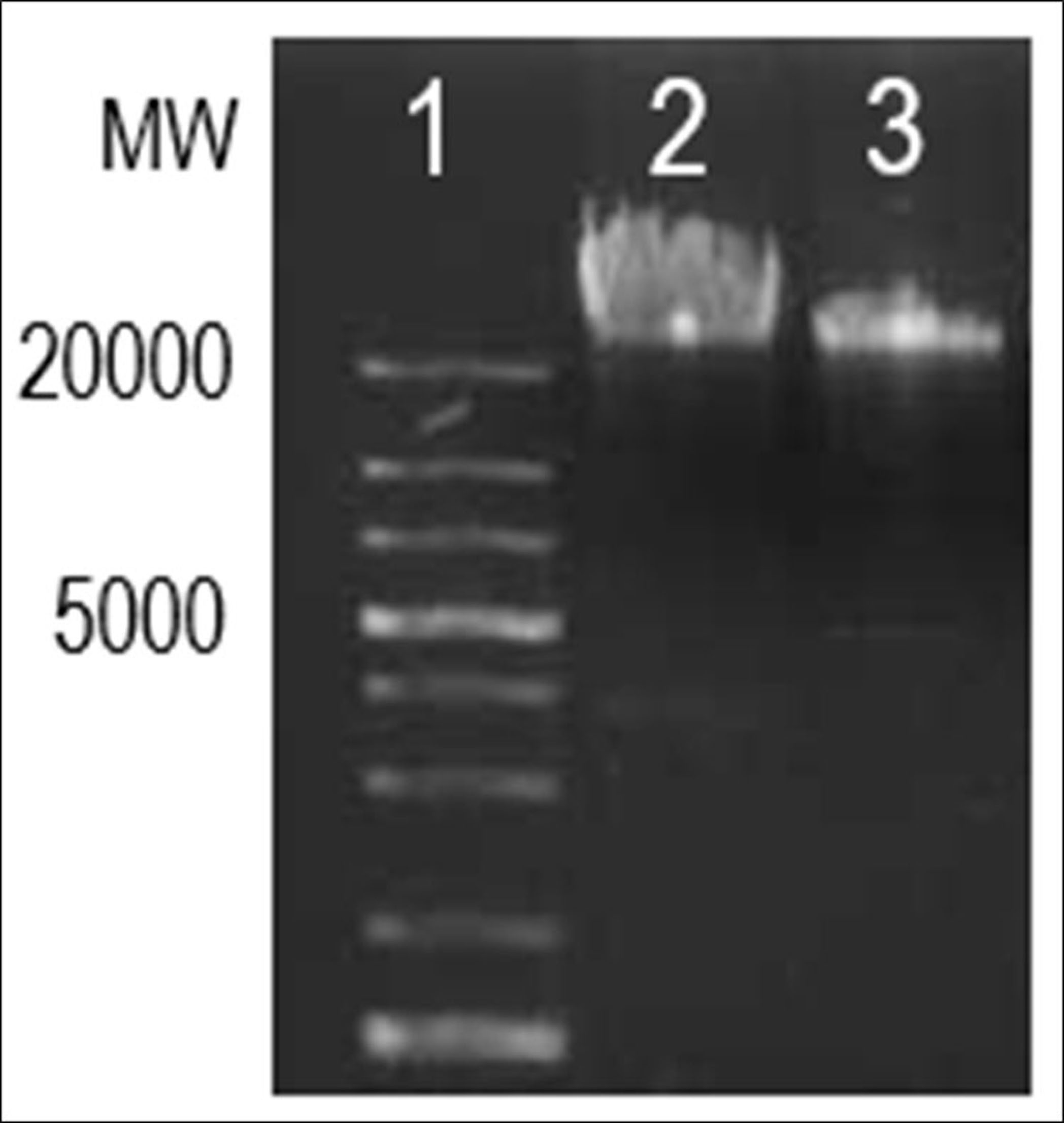
Fig. 1. gDNA of BSN3 and BMIC4 on 0.8% agarose gel electrophoresis; lane 1-1 kb plus DNA ladder (marker, bp); lane 2- B. subtillis Natto3; lane 3- B. methylotrophicus IC4
Whole genome sequencing of BSN3 and BMIC4 and related analysis
The whole genome sequence was performed using Illumina HiSeq 2500. Total paired reads were de novo assembled using MaSuRCA, SPades, Velvet and SOAP denovo2. The tRNA genes were predicted by tRNAscan-SE. Genes were finally predicted from the SPades and MaSuRCA assembled contigs (owing to the consistency in the results) for BSN3 and BMIC4 respectively, using Glimmer and annotated using BLASTX and CANOPI (Contig Annotator Pipeline). Phylogenetic tree was prepared using online NCBI tool phyloT. PFAM analysis for BSN3 and BMIC4 was carried out through the pfam database using Accession no. NC_000964 for B. subtilis and ABS72822 for B. methylotrophicus. Gene sequence similarity between BSN3 and BMIC4 was carried out through Needle (EMBOSS) which creates an optimal global alignment of two sequences using the Needleman-Wunsch algorithm.
Accession numbers of BSN3 and BMIC4
This WGS data for BSN3 and BMIC4 is submitted to ENA database under the accession number PRJEB31748 and PRJEB32025 respectively.
Comparison of BSN3 and BMIC4 genomes, in a nutshell
Whole genome sequencing is recognized as a phenomenal tool to capture the sequence of information on various organisms which can be correlated with respect to the variation in the genome and related phenotypes23. Key feature of the respective whole genome assemblies of BSN3 and BMIC4 like genome size, predicted number of genes, tRNA genes, average size of the genes and number of putative coding sequences with no matches (Fig. 1) showed remarkable difference (Table 1). BMIC4 genome was found to be larger than BSN3 with a delta of 613,629 bp. The genome size of BMIC4 was found to be bigger than the recently reported genomes of B. methylotrophicus strains9-10. Difference in the size of the genomes could be due to the expansion or reduction in the gene families. In general, prokaryotic genomes are considered to be dynamic and may increase or decrease in size via horizontal gene transfer and gene loss, respectively over the period of time due to evolution24-25. However, it is also reported that the chromosome alterations and reduction in the genome size is an uncommon phenomena in B. subtilis23 and B. methylotrophicus which is a recently discovered species1.
Table (1):
Key features of the whole genome sequencing of PGA producing novel Bacillus spp.
Parameters |
B. subtilis Natto3 |
B. methylotrophicus IC4i |
|---|---|---|
Genome size |
3,601,055 bp |
4,214,684 bp |
Assembled of contigs |
278 |
203 |
G+C content |
44.88 % |
43.74% |
Average gene length |
843.33 bp |
819.82 bp |
Total coding sequences/predicted genes |
3913 |
4341 |
Coding sequences with no matches |
57 |
117 |
Plasmid |
Not detected |
Not detected |
tRNAs |
52 |
74 |
ENA accession |
PRJEB31748 |
PRJEB32025 |
Number of genes with a significant BlastX hit |
3856 |
4223 |
i Recently communicated as B. methylotrophicus IC2 15
In our analysis, we also found that, both the bacteria were devoid of plasmid. In the current study, the average gene length of both the species was observed to be similar; however the total number of predicted genes was found to be more in BMIC4 due to the larger genome size. The number of the predicted gene of the BMIC4 was 4341 and the number of the most significant matched protein hits with the Uniprot database was 4223. In BSN3, number of the predicted genes and number of the most significant matched protein hits were 3913 and 3856 respectively. A thorough investigation of the pfam database revealed the presence of 2369 common protein families in both BSN3 and BMIC4. Surprisingly, BMIC4 being larger in genome size and more in gene number with significant BlastX hits showed smaller number of uncommon pfam (62) that were unique to BMIC4 compared to 389 unique pfam of BSN3 (Fig. 3, Table S1). The large genome of BMIC4 also attributes to a significant number of 117 genes which did not show any BlastX match. As previously discussed, B. methylotrophicus is newer species and due to the limited knowledge about un-annotated genomic segments and related regulatory mechanism, the need of the further details is essential.
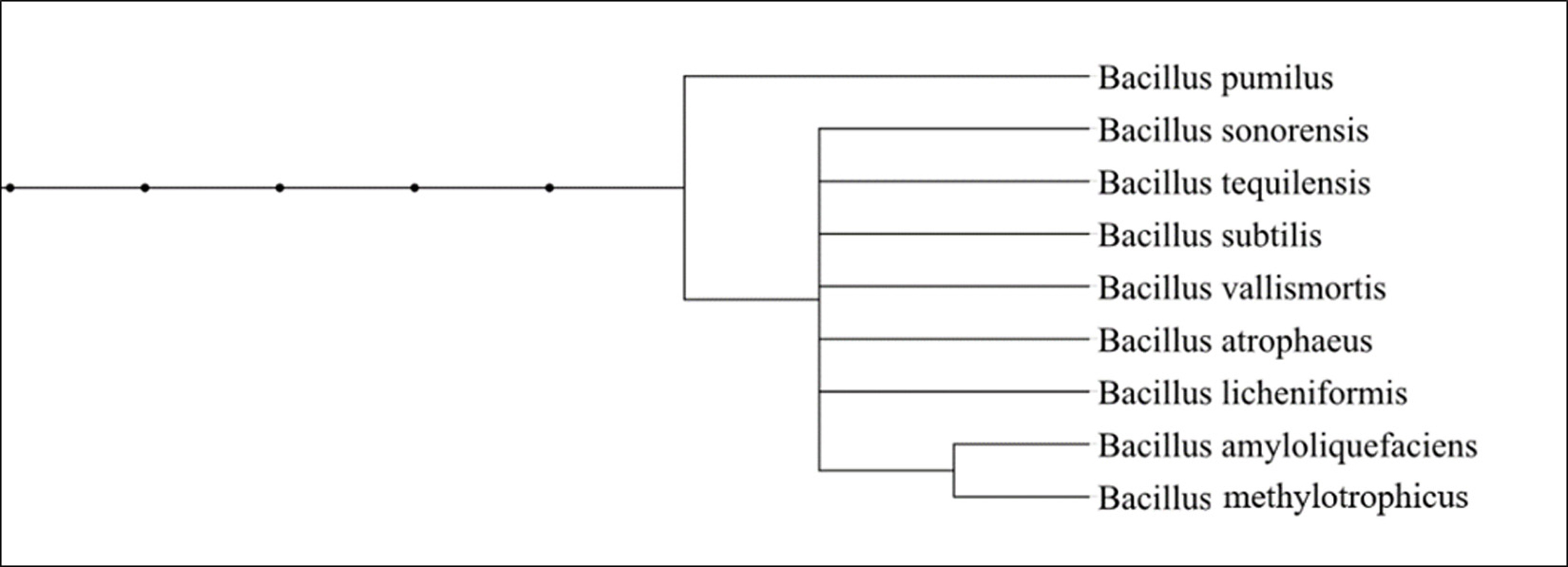
Fig. 2. Phylogenetic tree construction; fig. 2 shows the phylogenetic relationship which is based on 16S rDNA gene sequences from related species of the Bacillus genus
Sequence comparison analysis
PGA biosynthesis is a result of poly-merization of glutamic acid which is either incorporated in the media exogenously or glutamic acid derived intracellularly from tri-carboxylic acid cycle (TCA)5,7,26. The dependency of Bacillus species has been categorically divided into two types; Glutamic acid dependent and glutamic acid independent11. Other than the presence of this substrate, both BSN3 and BMIC4 were also found to have different nutritional requirement for PGA production which can directly be correlated with their genetic make-up. BMIC4 could grow well and produce PGA in presence of mono-sodium glutamate (MSG), peptone and mannitol. However, in these conditions, BSN3 failed to produce PGA15 which otherwise can biosynthesize PGA when glutamic acid and glucose is provided in the fermentation media, exogenously7. Glutamic acid poly-merization into PGA is facilitated via membrane associated PGA synthetase complex. This complex, in Bacillus spp. is coded by pgs operon comprising of the genes ywsC and ywtABC (also known as pgsBCAE or capBCA operon)4-5. A comprehension of the genetics in one strain can be rationalized to the other species but the comparative studies lead to understanding of deeper acumen about the internal system of the bacteria. As discussed above BSN3 and BMIC4 both produce PGA with different media compositions. This attracted more probing into the mannitol utilizing genes apart from pgs operonic genes as the former were important to study in BMIC4 strain with respect to PGA production. The insights on the genetic variation between BSN3 and BMIC4 strains of the aforementioned genes and their associated regulatory genes were thus found to be worth exploring from the nutritional preference perspective which ultimately may be indirectly responsible for the chain length of PGA.
PGA operonic gene cluster
We carried out the gene sequence similarity analysis of PGA producing genes and the results of the alignment are shown in Table 2. The sequences of all the PGA operonic genes were found to be 100% identical in both the bacteria except for pgsA (or ywtB). PgsA protein is made up of positively charged amino acids and is highly hydrophobic in nature. It has also been categorized as A1-type anchor protein found on the cell surface which may play a role in holding the PGA synthetase assembly to the cell membrane. The probable function of the product of pgsA may be that of a PGA transporter and seem to be involved in the elongation of PGA. It is suggested that the PgsA protein properties play a crucial role in removing the negatively charged PGA molecule from the active site of the PGA synthetase complex thereby helping in the extension of PGA molecule27-28. Gene disruption study by Urushibata et al showed that B. subtilis IFO16449 cells in which ywtB (homolog of pgsA) gene was absent, could produce PGA with reduced productivity, whereas cells with disrupted ywsC (homolog of pgsB) and ywtA (homolog of pgsC) could not produce PGA. This suggested that the gene ywsC and ywtA are essential for PGA production but ywtB may be required for maximising the PGA production4. In contrast to this, Ashiuchi et al reported that glutamate-dependent ATPase reaction was observed only in PgsBCA and PgsBC complexes. This study also showed that mutant B. subtilis ISW1214 cells without pgsA gene, could not produce PGA which implied that each constituent of the PgsBCA machinery was critical for PGA production29. In our previous study, we have reported an up-regulation of ywsC and ywtA genes and down-regulation of ywtB and ywtC genes simultaneously in the presence of glucose and glutamic acid. In the same study we observed PGA production of ~5 g/L which also indicated towards the critical role of mainly ywsC and ywtA genes7. The ambiguity that which components of pgsBCAE play the central role in PGA production has recently been reported by Sawada et al The same study concluded that recombinant B. subtilis expressing pgsBC gene on a vector without pgsA gene, produced 26 g/L PGA, making pgsBC genes essential for PGA production28. On the similar line, a recent review article by Hsueh et al, showed a phylogenetic analysis on the diversity of the pgs operonic genes, among few hundred representative strains from the Firmicutes phylum. The authors concluded that pgsC was the most conserved gene of the pgs operon across Bacillus spp. hence is irreplaceable. However, pgsB, pgsA and pgsE had sequence variations and may not affect the PGA synthesis12. Although these phylogenetic relationships of pgs operonic genes contradict the finding of Sawada et al who suggested that pgsB is an essential component for over-production of PGA, however both studies concur on inessentiality of pgsA. In the current study, pgsA (or ywtB) showed only 30% sequence similarity between BSN3 and BMIC4, which may imply that although the sequence similarity of pgsC is very low between these two species but may not have any impact on PGA production as the role of pgsA in PGA production is highly disputed. Further studies will be required to elucidate the role of pgsA gene in PGA production.
Table (2):
Percent similarity of various genes involved in the PGA production, degradation and regulation between BSN3 and BMIC4
| % similarity between BSN3 and BMIC4 | PGA producing genes | PGA degrading genes | Mannitol utilization genes |
PGA regulatory genes | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| pgsB | pgsC | pgsA | pgsE | pgdS | ggt | mtlF | mtlD | mtlR | mtlA | comP | comQ | comA | degU | degQ | |
| 100 | 100 | 30.4 | 100 | 100 | 41.7 | 100 | 100 | 100 | 100 | 47.9 | 38.1 | 100 | 99.4 | 100 | |
Mannitol utilization genes
In B. subtilis, mannitol utilization operon is comprised of mtlA, mtlF and mtlD genes (mtlAFD) and regulated by the product of mtlR. The gene mtlA codes for EIICBMtl, and the gene mtlF codes for EIIAMtl which comprises mannitol phosphoenolpyruvate (PEP):carbohydrate phosphotransferase system (PTS). Using this system mannitol is transported inside the cell and phosphorylated for further reactions. mtlD encodes for mannitol 1-phosphate 5-de-hydrogenase which converts the mannitol-1-phospahate to fructose-6-phospahate30 which can then enter the glycolysis. In our one of the previous studies, BMIC4 exhibited high PGA production with media containing mannitol as whereas BSN3 could not grow at all in the presence of 50 g/L mannitol15.
MtlR, the regulatory protein of mtlAFD operon contains two PTS regulatory domains, one EIIBGat– and one EIIAMtl-like domain. These regulatory domains possess potential PTS phosphorylation sites. A study showed that replacement of Histidine-342 or Histidine-399 with alanine, in PTS regulatory domain 2 blocked the phosphorylation of B. subtilis MtlR leaving it non-functional in vivo31. In this study, we found a 100% sequence similarity for mtlAFD and mtlR genes between BSN3 and BMIC4 hence there is no doubt on the non-functionality of the MtlR in BSN3. So the possibility that mutation in MtlR may have left it non-functional thereby no growth on mannitol was observed, is ruled out. This may imply to certain speculation that mtlAFD operon is non-functional in BSN3. Other possibility could be that its transcription may be attenuating through RNA binding proteins or some other factors in BSN3 which is known to exist for operons like sac, trp, and pyr of B. subtilis32. Further studies will be required to substantiate this possibility in BSN3.
Genes related to regulation of PGA production
Cell density-dependent phenotypes in B. subtilis such as the degradative enzymes synthesis, secondary metabolite production and the development of genetic competence are controlled by the quorum-sensing system (QS) ComQXPA.
PGA operon is also regulated by ComP-ComA signal transduction system and DegS-DegU two component system, which is very well understood6,12. At high cell density, ComP drives a series of phosphorylation reaction for ComA, DegQ and DegU proteins. Ultimately, phosphorylated DegU protein in turn regulates the expression of pgs operon6,12,33. In our sequence analysis study, we found out that the difference in nucleotide sequence of the genes namely comA, degU and degQ; between BSN3 and BMIC4 was barely any (100%, 99.4% and 100% respectively) whereas the sequence similarity of comP and comQ was below 50% (Table 2). The genes comP and comQ, are part of comQXP locus. In B. subtilis isolates or Bacillus pherotypes, high polymorphism has already been reported within this locus6,34-35. This polymorphism has arisen the question on the existence of phenomena of QS diversity, underlying mechanism and evolutionary significance which is yet to be investigated14. Despite these variations, the functionality of ComP protein may not be disturbed as it is reported that ComP protein of B. subtilis origin contains two conserved intracellular motifs which are histidine kinase domain (PFAM: PF07730) and an ATP-binding domain (PFAM: PF02518)36. Hence it is most likely that the variation in nucleotide sequence of comP exist outside these conserved domains and thereby not affecting the functionality of the protein. Since presence of QS is not only restricted to only B. subtilis and is found to exist across the Firmicutes phylum hence this machinery may also have role to play in regulating PGA production in BMIC437.
Genes related to PGA degradation
Primarily, ggt and pgdS (homolog of ywtD) are the genes which are involved in PGA degradation in Bacillus spp.12. The gene ggt codes for g-glutamyltransferase (EC 2.3.2.2; GGT) having an exo-g-glutamyl hydrolase action. On the other hand pgdS codes for g-DL-glutamyl hydrolase which may be associated to the DL-endopeptidase II family and is located right after pgs operon38-39. Various gene knock out studies with respect to ggt and pgdS have also shown increased PGA yield12. Between the genomes of BSN3 and BMIC4, the sequence similarity of “ggt” gene was merely ~41% (Table 2). These differences in the genomes of the both these Bacillus spp. very well correlates with the corresponding distance in their phylogenetic relationship (Fig. 2). Owing to the minor differences obtained in morphological, physiological, chemotaxonomic, phylogenetic and comparative genomic analysis of certain strains, it was recently proposed that B. methylotrophicus should be renamed as B. velezensis40. As discussed above, according to a recent review of Hsueh et al a phylogenetic analysis of many representative strains from the phylum Firmicutes revealed that pgdS gene was broadly absent in many Bacillus spp. including B. methylotrophicus and B. velezensis. If B. methylotrophicus could be the synonym of B. velezensis, then the probability of the pgdS gene not being found in various strains of both the Bacillus spp was very high12. On the contrary to the findings of Hsueh et al we report the presence of pgdS (ywtD) gene in the genome of BMIC4 and its sequence matched completely to BSN3. However, the mere presence of pgdS in the genome of BMIC4 does not confirm the functionality of this gene. In one of our latest study BMIC4 produced PGA in a range of ~1800-2300 kDa in the conditions tested without any genetic modification Whereas, a study in which genetically engineered B. amyloliquefaciens with deleted pgdS gene also produced PGA with improved yield but molecular weight was very low compared to our study26 Given the fact that both these studies had different media compositions as well as the culture strain, yet BMIC4 may prove to be a potent novel strain for PGA production using mannitol, MSG and peptone as substrates. Molecular weight of PGA produced by BSN3 is in a range of ~1200-1400 kDa (unpublished data). The rationale behind it may be that both the genes; ggt and pgdS are functional39-40. As we encountered the presence of pgdS gene in BMIC4, we also dropped the chances of rationalizing the PGA production with high molecular weight. Still, this opens up the avenues to carry out further comparative expression studies of the genes with respect to the PGA production and degradation. This may correlate the functionality of pgdS and high molecular weight of PGA produced by BMIC4.
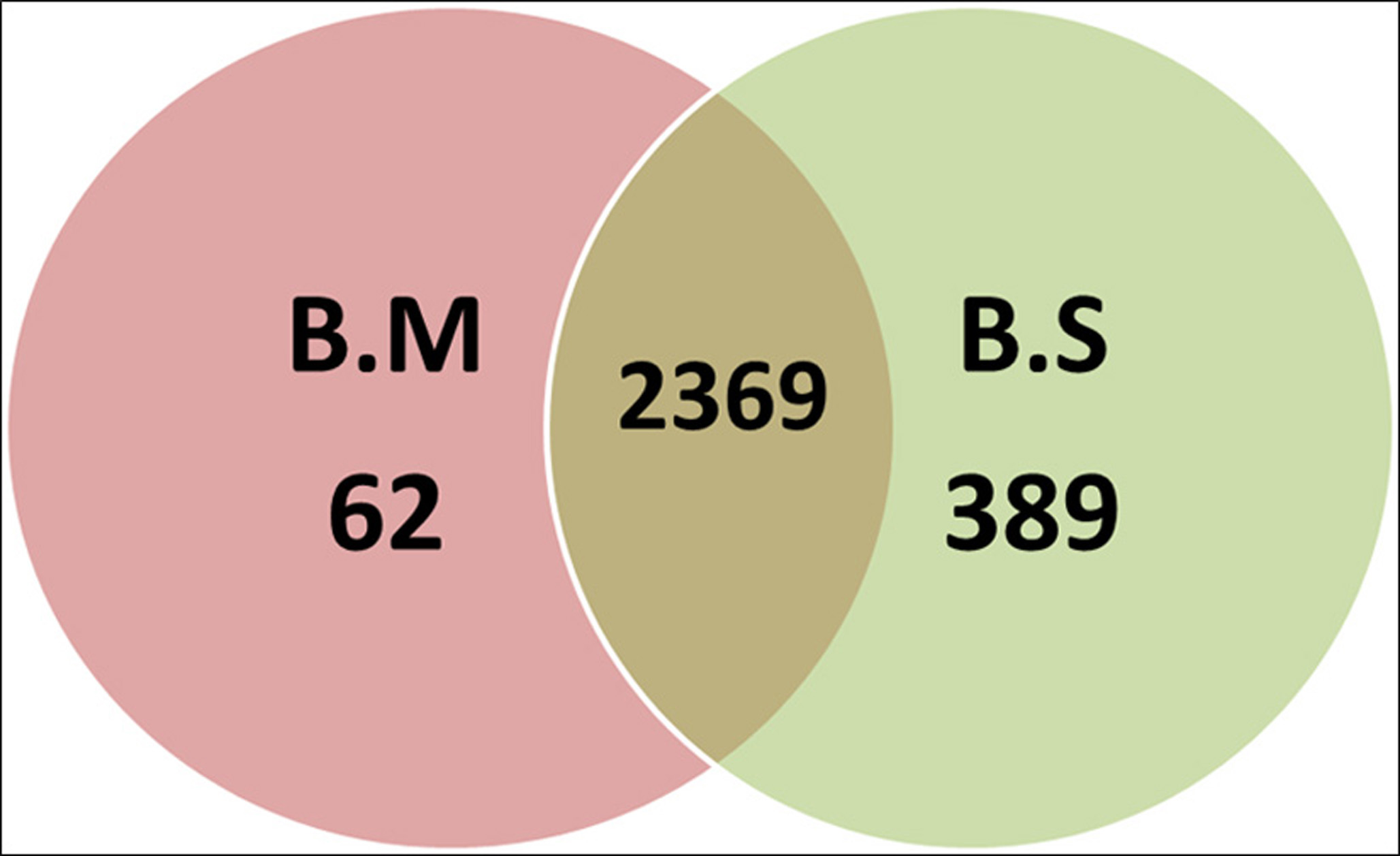
Fig. 3. Venn diagram for the number of unique and common protein domain of BSN3 and BMIC4 based on pfam database
BSN3 and BMIC4 as reported in our previous studies are the novel PGA producers and differ greatly with respect to nutrient and substrate utilization, growth, and PGA production which was evident from their unique expression pattern of pgs operon genes. A comparative study of the whole genome sequence of both the organisms was carried out for PGA producing and regulatory genes. BMIC4 genome was larger than BSN3 with more number of predicted genes and proteins. Major difference in genes was found in the pgsA (ywtB) and ggt which are involved in PGA production and degradation respectively. The role of pgsA in PGA production is widely disputed5,7,28 hence the huge differences in the sequence similarity of pgsA may not be critical. pgdS has been broadly found to be absent in many strain of B. methylotrophicus and B. velezensis and present in B. subtilis12. In our analysis we report the presence of pgdS in BMIC4. On the similar line, we propose relevant expression studies which could throw light on the functionality of pgdS product from B. methylotrophicus origin. Due to limited information on the genome of B. methylotrophicus, this comparative study will provide understandings on the comparative genomic variations with B. subtilis found in the genome of BMIC4 compared to B. subtilis. Another way forward could be to indulge into the genomic diversity of B. methylotrophicus to draw critical insights on the genome dynamics across strains.
Acknowledgements
This work was financially supported by Tata Chemicals Limited, Pune, India. DPT and PMC are recipients of Prime Minister’s Fellowship Scheme for Doctoral Research, a public-private partnership between Science & Engineering Research Board, Department of Science & Technology, Government of India, and Confederation of Indian Industry. Whole genome sequencing was carried out at AgriGenome Labs Private Limited, Kochi (Kerala, India). Authors thank Manipal University for registering DPT and PMC as Ph.D. scholars to pursue their research work on poly--glutamic acid. This research did not receive any specific grant from funding agencies in the public/commercial/not-for-profit sectors.
Conflict Of Interest
The authors declare that there is no conflict of interest.
Authors’ Contributions
DT, PC, RR and AD conceptualized the study. DT, PC, AB and AD contributed in data interpretation. DT, NU, AB and AD designed the figures and tables. DT compiled information from the results and literature and drafted the manuscript. PC helped in draft improvisation. AD, RR and AB supervised and reviewed the manuscript. DT, PC, NU, AB, RR and AD read and approved the manuscript.
Funding
None.
Data Availability
The datasets generated and analysed during the current study of Whole Genome Sequencing for Bacillus subtilis Natto3 and Bacillus methylotrophicus IC4 is submitted to ENA database under the accession number PRJEB31748 and PRJEB32025 respectively.
Ethics Statement
Not applicable.
- Ogunleye A., Bhat A., Irorere V.U., Hill D., Williams C., Radecka I. Poly-g-glutamic acid: production, properties and applications. Microbiology, 2015; 161(Pt 1):1-17.
- Park C., Choi Y.H., Shin H.J., Poo H., Song J.J., Kim C.J., Sung M.H. Effect of High-Molecular-Weight Poly-g-gamma $-Glutamic Acid from Bacillus subtilis (chungkookjang) on Ca Solubility and Intestinal Absorption. J. Microbiol. Biotechnol., 2005; 15(4): 855-858.
- Park J.H., Choi J.C., Sung M.H., Kang J.H., Chang M.J. High molecular weight poly-gamma-glutamic acid regulates lipid metabolism in rats fed a high-fat diet and humans. J. Microbiol. Biotechnol., 2011; 21 (7): 766-775.
- Urushibata Y., Tokuyama S., Tahara Y. Characterization of the Bacillus subtilis ywsC gene, involved in g-polyglutamic acid production. J. Bacteriol., 2002; 184(2): 337-743.
- Ashiuchi M. Microbial production and chemical transformation of poly g glutamate. Microb Biotechnol, 2013; 6(6):664-674.
- Tran L.S., Nagai T., Itoh Y. Divergent structure of the ComQXPA quorum sensing components: molecular basis of strain specific communication mechanism in Bacillus subtilis. Mol. Microbiol., 2000; 37(5): 1159-1171.
- Tiwari D.P., Chatterjee P.M., Rotti H., Chand B., Raval R., Dubey A.K. Expression dynamics of the poly-g-glutamic acid biosynthesis genes of Bacillus subtilis in response to glucose and glutamic acid-a pilot study. FEMS Microbiol. Lett., 2018; 365(22):fny248.
- Kunst F., Ogasawara N., Moszer I., Albertini A.M., Alloni G.O., Azevedo V., Bertero M.G., Bessieres P., Bolotin A., Borchert S., Borriss R. The complete genome sequence of the gram-positive bacterium Bacillus subtilis. Nature, 1997; 390(6657): 249.
- Gerbore J., Brutel A., Lemainque A., Mairey B., Mיdigue C., Vallenet D., Lefort F., Grizard D. Complete genome sequence of Bacillus methylotrophicus strain B25, a potential plant growth-promoting rhizobacterium. Genome Announc., 2016; 4(2): e00058-16.
- Liu B., Ge B., Azhar N., Zhao W., Cui H., Zhang K. Complete Genome Sequence of Bacillus methylotrophicus Strain NKG-1, Isolated from the Changbai Mountains, China. Genome Announc., 2018; 6(3):e01454-17.
- Peng Y., Jiang B., Zhang T., Mu W., Miao M., Hua Y. High-level production of poly (g-glutamic acid) by a newly isolated glutamate-independent strain, Bacillus methylotrophicus. Process Biochem, 2015; 50(3): 329-335.
- Hsueh Y.H., Huang K.Y., Kunene S., Lee T.Y. Poly-g-glutamic acid synthesis, gene regulation, phylogenetic relationships, and role in fermentation. Int. J. Mol. Sci., 2017; 18(12): 2644.
- Madhaiyan M., Poonguzhali S., Kwon S.W., Sa T.M. Bacillus methylotrophicus sp. nov., a methanol-utilizing, plant-growth-promoting bacterium isolated from rice rhizosphere soil. Int. J. Syst. Evol. Microbiol., 2010; 60(10): 2490-2495.
- Oslizlo A., Stefanic P., Vatovec S., Beigot Glaser S., Rupnik M., Mandic Mulec I. Exploring ComQXPA quorum sensing diversity and biocontrol potential of Bacillus spp. isolates from tomato rhizoplane. Microb Biotechnol, 2015; 8(3): 527-540.
- Chatterjee P.M., Datta S., Tiwari D.P., Raval R., Dubey A.K. Selection of an effective indicator for rapid detection of microorganisms producing g-polyglutamic acid and its biosynthesis under submerged fermentation conditions using Bacillus methylotrophicus. Appl. Biochem. Biotechnol., 2018; 185(1): 270-288.
- Liu B., Ge B., Azhar N., Zhao W., Cui H., Zhang K. Complete Genome Sequence of Bacillus methylotrophicus Strain NKG-1, Isolated from the Changbai Mountains, China. Genome Announc., 2018; 6(3):e01454-17.
- Earl A.M., Losick R., Kolter R. Ecology and genomics of Bacillus subtilis. Trends Microbiol., 2008; 16(6): 269-275.
- Mandic-Mulec I., Prosser J.I. Diversity of endospore-forming bacteria in soil: characterization and driving mechanisms, 2011, pp. 31-59. In Logan N., Vos P. (eds) Endospore-forming Soil Bacteria. Soil Biology. Springer, Berlin, Heidelberg.
- Sirisansaneeyakul S., Cao M., Kongklom N., Chuensangjun C., Shi Z., Chisti Y. Microbial production of poly-g-glutamic acid. World J. Microbiol. Biotechnol., 2017; 33(9):173.
- Cai D., He P., Lu X., Zhu C., Zhu J., Zhan Y., Wang Q., Wen Z., Chen S. A novel approach to improve poly-g-glutamic acid production by NADPH regeneration in Bacillus licheniformis WX-02. Sci. Rep., 2017; 7: 43404.
- Goto A., Kunioka M. Biosynthesis and Hydrolysis of Poly(y-glutamic acid) from Bacillus subtilis IF03335. Biosci. Biotech. Biochem., 1992; 56: 1031-1035.
- Ko Y.H., Gross R.A. Effects of glucose and glycerol on g-poly-(glutamic acid) formation by Bacillus licheniformis ATCC 9945a. Biotechnol. Bioeng., 1998; 57: 430-437.
- Sharma A., Satyanarayana T. Comparative genomics of Bacillus species and its relevance in industrial microbiology. Genomics insights, 2013; 6: GEI-S12732.
- Pushker R., Mira A., Rodrםguez-Valera F. Comparative genomics of gene-family size in closely related bacteria. Genome Biol., 2004; 5 (4): R27.
- Brito P.H., Chevreux B., Serra C.R., Schyns G., Henriques A.O., Pereira-Leal J.B. Genetic competence drives genome diversity in Bacillus subtilis. Genome Biol. Evol., 2017; 10(1): 108-124.
- Feng J., Gu Y., Quan Y., Cao M., Gao W., Zhang W., Wang S., Yang C., Song C. Improved poly-g-glutamic acid production in Bacillus amyloliquefaciens by modular pathway engineering. Metab. Eng., 2015; 32: 106-115.
- Ashiuchi M., Nawa C., Kamei T., Song J.J., Hong S.P., Sung M.H., Soda K., Yagi T., Misono H. Physiological and biochemical characteristics of poly-g-glutamate synthetase complex of Bacillus subtilis. Eur. J. Biochem., 2001; 268(20): 5321-5328.
- Sawada K., Araki H., Takimura Y., Masuda K., Kageyama Y., Ozaki K., Hagihara H. Poly-L-gamma-glutamic acid production by recombinant Bacillus subtilis without pgsA gene. AMB Express, 2018; 8(1): 110.
- Ashiuchi M., Shimanouchi K., Horiuchi T., Kamei T., Misono H. Genetically engineered poly-g-glutamate producer from Bacillus subtilis ISW1214. Biosci. Biotechnol. Biochem., 2006; 70(7): 1794-1797.
- Heravi K.M., Altenbuchner J. Regulation of the Bacillus subtilis mannitol utilization genes: promoter structure and transcriptional activation by the wild-type regulator (MtlR) and its mutants. Microbiology, 2014; 160(1): 91-101.
- Joyet P., Derkaoui M., Poncet S., Deutscher J. Control of Bacillus subtilis mtl operon expression by complex phosphorylation dependent regulation of the transcriptional activator MtlR. Mol. Microbiol., 2010; 76(5): 1279-1294.
- Yanofsky C. Transcription attenuation: once viewed as a novel regulatory strategy. J. Bacteriol., 2000; 182: 1-8.
- Stanley, N.R.; Lazazzera, B.A. Defining the genetic differences between wild and domestic strains of Bacillus subtilis that affect poly-g-DL-glutamic acid production and biofilm formation. Mol. Microbiol., 2005; 57: 1143–1158.
- Ansaldi M., Marolt D., Stebe T., Mandic Mulec I., Dubnau D. Specific activation of the Bacillus quorum sensing systems by isoprenylated pheromone variants. Mol. Microbiol., 2002; 44(6): 1561-1573.
- Stefanic P., Mandic-Mulec I. Social interactions and distribution of Bacillus subtilis pherotypes at microscale. J. Bacteriol, 2009; 191(6): 1756-64.
- Piazza F., Tortosa P., Dubnau D. Mutational analysis and membrane topology of ComP, a quorum-sensing histidine kinase of Bacillus subtilis controlling competence development. J. Bacteriol., 1999; 181(15): 4540-4548.
- Dogsa I., Choudhary K.S., Marsetic Z., Hudaiberdiev S., Vera R., Pongor S., Mandic-Mulec I. ComQXPA quorum sensing systems may not be unique to Bacillus subtilis: a census in prokaryotic genomes. PloS one, 2014; 9(5): e96122.
- Kimura K., Tran L.S., Uchida I., Itoh Y. Characterization of g-glutamyltransferase and its involvement in the degradation of capsule poly-g-glutamate. Microbiology, 2004; 150(12):4115-23.
- Suzuki T., Tahara Y. Characterization of the Bacillus subtilis ywtD gene, whose product is involved in γ-polyglutamic acid degradation. J. Bacteriol., 2003; 185(7):2379-82.
- Dunlap, C.A., Kim, S.J., Kwon, S.W., Rooney, A.P. Bacillus velezensis is not a later heterotypic synonym of Bacillus amyloliquefaciens; Bacillus methylotrophicus, Bacillus amyloliquefaciens subsp. plantarum and ‘Bacillus oryzicola’ are later heterotypic synonyms of Bacillus velezensis based on phylogenomics. Int. J. Syst. Evol. Microbiol., 2016; 66(3):1212-1217.
© The Author(s) 2019. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.