


ISSN: 0973-7510
E-ISSN: 2581-690X
This research explores bacterial communities in individuals diagnosed with oral cancer, comparing them to healthy individuals to identify potential variations associated with the condition. The study involved collecting 40 swabs from oral cancer patients, post-therapeutic patients, and healthy individuals, amplifying DNA samples, processing raw data using Perl scripts and Prinseq Lite, performing metagenomic analysis using QIIME 2-2022.2, and taxonomic classification using Greengenes2. There are 91.89% of good quality sequences for downstream analysis. Analysis data indicates that individuals who suffer from oral cancer had much higher prevalence of phylum Actinobacteriota, Firmicutes_A, Campylobacterota, Fusobacteriota, and Patescibacteria. Total 298 species identify in current study, among this Leptotrichia (0.0015%), Prevotella (0.0041%), and Capnocytophaga (0.0052%) are predominant in oral cancer patients compare to healthy individuals. 23 species are absent in normal individuals and post-therapeutic patients but are dominant in oral cancer patients. The increased occurrence suggests a link between this group of bacteria and oral cancer. By comparing the abundance of alpha and beta microorganisms in patients with oral cancer to those in good health, the study highlights the importance of the oral microbial community in maintaining health and preventing disease. It also studies how habits like tobacco use affect microbial communities and how they can raise the risk of disease. In cancer patients, oxidative stress and glycolysis are enhanced, and while certain metabolic abnormalities recovered after therapy, many remain, showing the long-term impact of the illness and treatment. These data suggest that post-treatment microbial regeneration may not occur, increasing cancer recurrence risk. The study’s finding of microbial biomarkers, particularly those related to dysbiosis and changed tumor microenvironment, may inform oral cancer prognostic, therapeutic, and diagnostic methods. This metagenomic work contributes to a better understanding of how lifestyle factors influence microbial ecosystems, allowing lifestyle adjustments to lessen health risks associated with changes in microbial populations.
Bacterial Diversity, Dominant Bacteria, Oral Cancer, Metagenomics
Cancer is a significant public health concern in both industrialized and developing nations globally. Globally, an annual incidence of over 450,000 new cases of oral cancer is documented, with a survival rate of about 40% to 50% within 5 years following diagnosis. Interestingly, a large fraction of these cases, specifically two-thirds-occur in Asian nations like Bangladesh, India, Indonesia, Pakistan, and Sri Lanka.1,2 There is a major variation in the prevalence of oral cancer between India and Western countries, with a significant proportion of cases, around 70%, being documented in advanced stages. The five-year survival rates are estimated to be under 20%, primarily attributed to the very low and practically nonexistent likelihood of achieving a treatment once the disease is detected in its advanced stages.3 Oral cancer is a malignancy that specifically targets the oral cavity and pharynx, encompassing the tongue, lips, gums, cheeks, and salivary glands. Chronic illness is a significant public health issue that can have profound and detrimental impacts on both people and their families.4,5 The etiology of oral cancer remains uncertain; nevertheless, some elements have been suggested as possible risk factors for its development. Oral cancer is known to be significantly influenced by tobacco use, which includes chewing betel nuts, smoking cigarettes, and using smokeless tobacco. These tobacco products expose users to the human papillomavirus (HPV), which is linked to the development of oral cancer. Additionally, Excessive alcohol consumption has been identified as an indicator of risk for the occurrence of oral cancer.4,6,7 A healthy bacterial microbiota and maintaining homeostasis between the human host and microbiota play a crucial role in ensuring optimal physiological functioning of the human body.8 In the environment of oral cancer, a diverse array of over 700 distinct bacterial species has been identified. The process of interaction involves the cooperative interactions of interdependent and harmful microorganisms to maintain equilibrium. Within the mouth cavity, where saliva has a pH profile ranging from 6.5 to 7.5 and a temperature of 37 °C, bacterial species exhibit a stable biological niche. In order to diagnose and track oral diseases and evaluate how they are progressing, saliva testing is essential.9 In contrast to eukaryotic cells, humans harbor a higher abundance of prokaryotic organisms. Prokaryotic microorganisms play a crucial role in allowing specific biological processes that humans are unable to perform individually, hence protecting against pathogenic microorganism infections. During the early 1990s, scientists had the belief that the human genome sequence would provide a comprehensive understanding of the fundamental factors contributing to human function and disease. However, the investigation of the human genome has been limited to a rudimentary examination of our cells’ genetic makeup.10 When compared to other physiological regions, it is observed that the oral microbiota exhibits the least amount of variation in beta diversity, while displaying the highest variation in alpha diversity. Furthermore, there is minimal variation in the composition of the oral cavity microbiome across individuals.11,12 Smoking among individuals is associated with a significant elevation in the likelihood of cancer development. Several studies have provided empirical support for the existence of more than 60 distinct carcinogenic chemicals inside tobacco smoke.13 After an intake of alcohol, the observable presence of alcohol on the surface of the tongue may endure for an extended amount of time. A uniform distribution of absorbed alcohol in bodily fluids leads to a corresponding reduction in the levels of ethanol in both gastric saliva and circulation. The specific processes via which alcohol consumption affects the composition of the oral microbiota are still not well understood. A hypothesis suggests that persons who have inadequate oral hygiene may encounter a rise in the production of microbial compounds that exhibit potential carcinogenic characteristics as a result of excessive bacterial proliferation. In this context, the microbial manufacture of acetaldehyde, a carcinogenic compound, from ethanol holds significant importance.14 The impact of tobacco on disease progression can be attributed to its propensity to alter the microbial communities within the oral cavity.15,16 A key factor in the development of mouth cancer is bacteria because they stop cells from dying, make them divide, help them invade, cause chronic inflammation, and make carcinogens.17 The emergence of inflammatory conditions and oral cancers has been linked to Prevotella species. The imbalance of the saliva microbiome caused by tobacco use in smokers, particularly the increased presence of Prevotella and Megasphaera species, may contribute to the advancement of several diseases.18 A total of 53 species showed a notably elevated prevalence among those who consume tobacco in comparison to those who identify as healthy. A collective of 15 species were found to be absent in individuals without any health conditions. However, these species were observed in notably greater quantities among those who partake in tobacco use and cigarettes. This study identifies 18 different types of Prevotella which have a notably elevated prevalence among people who engage in tobacco chewing and tobacco smoking, in comparison to people who are classified as normal and healthy.19 Whole genome sequencing has become more useful for the thorough examination of the genomes of a number of harmful microorganisms due to the expansion of knowledge about these organisms and the abundance of data regarding the diseases they cause.20 The report titled “The Emerging Field of Metagenomics: Unveiling the Enigmas of Our Microbial World” by the committee of US National Research Council emphasizes the transformative impact of metagenomics on microbiology research. This innovation has provided a valuable opportunity to explore the previously unexplored realm of microorganisms and their diverse characteristics.21 Over the course of the previous decade, several scholarly investigations have explored the diverse bacterial species associated with oral squamous cell carcinoma (OSCC) from novel perspectives. The many studies have revealed both similarities and differences in their results. The objective of this study was to use 16S rRNA amplicon sequencing to examine the genetic material of oral bacteria obtained from healthy individuals and those diagnosed with cancer. This study revealed a statistically significant association between oral microbiota and oral squamous cell carcinoma (OSCC). Microbiological diagnostics involve the identification and analysis of suspected pathogens present in clinical samples, offering necessary information for the efficient control and prevention of human illnesses.22 Traditional diagnostic approaches used in microbiology include several techniques such as cultivating microorganisms, conducting laboratory tests to identify antibodies or antigens specific to infections, and using PCR (polymerase chain reaction) to detect bacterial nucleic acids. These methods allow the identification and characterization of pathogenic microorganisms included in clinical samples.23 The field of metagenomics has recognized a significant rise in its significance as a novel approach for investigating microbiological phenomena, largely because of the current developments in DNA sequencing technology. Several recent research have employed a metagenomic methodology to investigate the functions of bacteria in oral hygiene disorders.24
This study aims to determine the possible bacterial populations that may be present in those who have been diagnosed with oral cancer. This study aims to investigate the significant variations in bacterial communities between individuals diagnosed with oral cancer and those who are in good health. By comparing these two groups, the study intends to emphasize the differences that could be linked to the occurrence of oral cancer.
Sample collection and DNA isolation
The study involved 40 male participants, including both patients and volunteers. Among them, 4 patients were in the 1st stage of oral cancer, 8 were in the 2nd stage, 8 were in the 4th stage, 12 were post-treatment patients, and 8 were healthy volunteers. Patients with HIV, diabetes, or autoimmune diseases were excluded from the study. The study was approved by the Sterling Hospital Ethics Committee in Ahmedabad, Gujarat, India (SHEC/HS/OC-Study/235-2022). The research aimed to recruit individuals with oral cancer who had a history of long-term tobacco use, as well as healthy individuals. Written consent was obtained from all participants prior to sample collection. Following the instructions provided by the manufacturer, DNA was isolated using the QIAamp DNA Mini Kit (Catalog number 51304) and 50 µL of elution buffer. After collection, the DNA was immediately used or stored at -20 °C until it was amplified using PCR.
16S rRNA amplification and bacterial metagenome sequencing
We utilized a primer set that precisely targets the V3-V4 hypervariable region of the bacterial 16S rRNA gene for the purpose of amplifying the gene and providing subsequent metagenome sequencing. An initial denaturation step at 95 °C for 5 minutes was performed at the beginning of the PCR procedure to guarantee thorough separation of the DNA strandsSubsequently, 31 amplification cycles were conducted, with each cycle including a denaturation step at 95 °C for 1 minute, an annealing step at 58 °C for 50 seconds, and an extension step at 72 °C for 1 minute. The amplification procedure was completed by a final extension at 72 °C for 7 minutes to ensure an accurate synthesis of all amplicons. A total volume of 25 µL was used to prepare the PCR reaction mixture. This includes 1 µL of each primer, prepared to a final concentration of 5 picomoles, and 12.5 µL of 2X KAPA HiFi HotStart Ready Mix, chosen for its excellent accuracy and durability in amplifying GC-rich regions. In order to improve the efficiency and accuracy of the amplification process, a quantity of 90-100 ng of template DNA was introduced, together with 1 µL of Bovine Serum Albumin (BSA). BSA is recognized for its ability to avoid the inhibition of PCR by binding to any inhibitors containing in the DNA samples. In order to enhance both the yield and specificity, a nested PCR was performed which utilized the original PCR result as the template. The PCR products obtained were subsequently seen and measured by electrophoresis on a gel containing 2% agarose dye. By the manufacturer’s instructions, the PCR products were purified by employing 20 µL of AMPure XP beads (Beckman Coulter; Catalog No. A63881) to remove any remaining primers, nucleotides, and enzymes. The extracted products were separated in 50 µL of a 10 mM Tris buffer (pH 8.5) to preserve the integrity of the DNA.25 The library was prepared by ligating Illumina adapters to the purified PCR products using the Nextera XT Index Kit V2 from Illumina®, USA; Catalog No. 15052164. An important phase in the DNA preparation for sequencing on the Illumina platform. Quantification of double-stranded DNA (dsDNA) was performed using the Qubit dsDNA HS Assay Kit (Thermo Fisher Scientific®, USA; Catalog No. 2438348) in combination with the Qubit Fluorometer 2.0 to ensure precise measurement for subsequent applications. Following ligation, the PCR products received further purification to exclude unligated adapters or primer dimers by treatment with AMPure XP beads at a 0.9X ratio. The Quantification of dsDNA was performed again using the Qubit dsDNA HS Assay Kit in order to verify the concentration and integrity of the library. The latter library was subsequently concentrated to a concentration of 4 nM and introduced into the flow cell at a concentration of 100 pM. Genomic sequencing was conducted on a NovaSeq 6000 SP equipment utilising a 500-cycle kit containing 250X2 base pair paired-end reads. The use of this high-throughput sequencing method facilitated thorough coverage of the bacterial 16S rRNA gene, therefore permitting rigorous taxonomic profiling of the bacterial populations present in the samples.25
Metagenomic bioinformatics and statistical analysis
In order to carry out quality filtering, trimming, and de-duplication of the raw sequencing reads acquired from the NovaSeq 6000 platform, Prinseq-lite, a Perl-based script, was initially used for processing. The first stage of preprocessing included eliminating bases of poor quality, reducing adapter sequences, and eliminating sequences that did not satisfy the minimal quality requirement, therefore ensuring the retention of only high-quality reads for subsequent analysis.26
Following the initial preprocessing, the filtered data was loaded into QIIME 2-2022.2 software to conduct a thorough investigation of the microbial community. Denoising and demultiplexing of the readings were performed using the DADA2 pipeline within QIIME 2. DADA2 is a robust platform that rectifies sequencing errors, eliminates chimeric sequences, and deduces precise amplicon sequence variants (ASVs) from the unprocessed sequencing data. This stage is essential as it enables a detailed examination of microbial diversity without the requirement to group readings into operational taxonomic units (OTUs). Utilizing the GreenGenes2 database, a carefully selected compilation of 16S rRNA gene sequences, the ASVs were mapped to recognized bacterial taxa, allowing for accurate identification and categorization of the microbial communities found in the samples. In-depth taxonomic analysis offered a comprehensive summary of the bacterial makeup among several sample groupings.26-28 Quantitative and integrative analysis of the microbiome data were conducted using MicrobiomeAnalyst 2.0. These tertiary analyses, such as differential abundance testing and diversity metrics, were conducted to identify significant differences in microbial composition between oral patients and healthy individuals.29-31
Per Sequence Quality Scores indicate the quality or reliability of each DNA base (nucleotide). The Per Sequence GC Content analysis, which has an average of 53%, investigates the guanine and cytosine nucleotides of DNA to uncover both functional and structural characteristics. Out of 7.50 Gbp raw data, 6.75 Gbp is good quality (96%). This good quality data is use for downstream analysis. The graph has reached the required sequencing depth and sample size to capture the total diversity of the microbiota, as indicated by the rarefaction curve, as these criteria have been satisfied. Every sample had a plateau in the alpha rarefaction curve when the sequencing depth hit 10,000 reads.
Diversity and structure of microbial communities across samples
In the current study, 40 samples were sequenced in duplicate using an Illumina MiSeq system, generating a total of 8136595 read counts. Following quality trimming and chimera checking, high-quality sequences with an average length of 245 bp were obtained for downstream analysis. The read counts per sample ranged from 136687 to 362,071, with an average of 203414. The Greengenes (GG) database was used to align and classify the sequences, producing 2108 distinct OTUs. Low abundance features were eliminated by using further low count and variance filters. After filtering stages, 1132 characteristics are still included in the Terteray analysis. These OTUs were subsequently divided into 101 genera and several phyla (Figure 1). Given that the Good’s estimation of coverage was 99.26%, it is likely that the bulk of the bacterial sequences found in the samples are represented by the 16S rRNA sequences found in this investigation. Figure 1 shows that twelve phyla were identified in all samples: Actinobacteriota, Bacteroidota, Campylobacterota, Firmicutes_A, Firmicutes_C, Firmicutes_D, Fusobacteriota, Patescibacteria, Proteobacteria, Spirochaetota, and Synergistota. Chloroflexota only found to be present in Cancer stage 4 and Desulfobacterota_I is absent in healthy; whereas, present in various stages of cancer as well as post therapetic individuals.

Figure 1. Phylogenetic tree and occurrence of phylum across samples
It was shown that the bacterial communities of cancer samples had a much higher Shannon diversity index (Figure 2) than the clinically normal control samples. The Shannon diversity index is a measure of microbial variety that takes into consideration both species richness and evenness. Both the Oral Cancer Stage-1 and Stage-4 patients (adjusted p-value = 0.0011942) and Normal Healthy Persons and Stage-4 patients (adjusted p-value = 0.001718) showed significant differences according to the Shannon index. This increase in diversity may be connected to the illness state and its effect on the local microbiota, since it indicates a more varied microbial habitat in the malignant tissues. Increased microbial diversity in cancer patients may be due to a changed immunological state or a more diverse range of bacterial species being supported by the tissue environment.
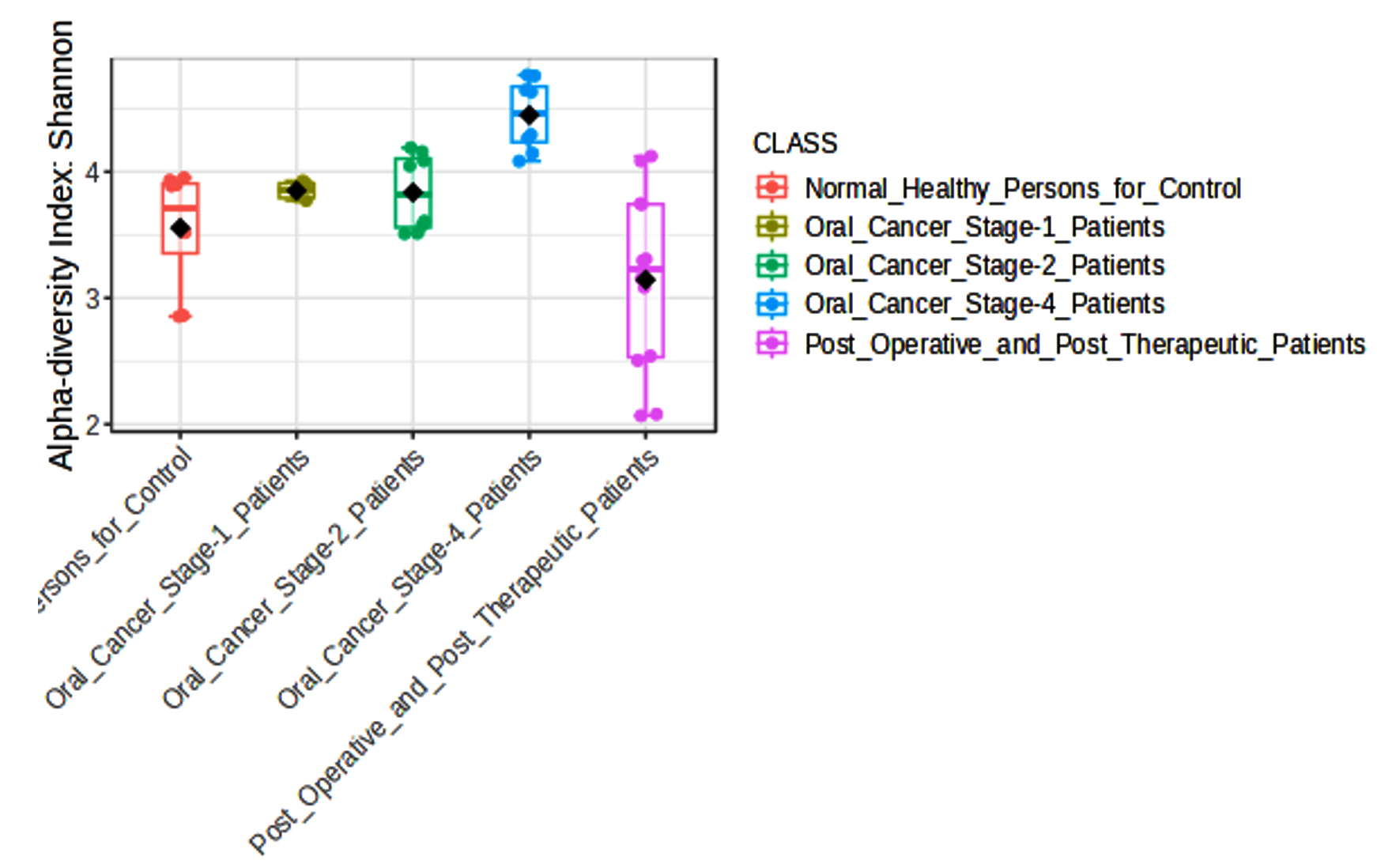
Figure 2. Box plot of the alpha diversity among groups
We evaluated variations in microbial communities by comparing different groups in our beta diversity study, as shown in Figure 3. Between Normal Healthy Persons and Oral Cancer Stage-1, Stage-2, and Stage-4 patients, the Bray-Curtis index showed significant differences (F-values ranging from 6.6165 to 8.189, adjusted p-values between 0.001 and 0.005). Jensen-Shannon divergence, which had F-values ranging from 8.0575 to 11.059 and adjusted p-values from 0.001 to 0.002, also showed significant differences in these comparisons. Weighted UniFrac analysis, with adjusted p-values ranging from 0.005 to 0.11167 and F-values ranging from 2.6966 to 7.7017, revealed significant findings largely between Oral Cancer Stage-1 and other groups. Together, our findings highlight significant changes in the composition of the microbial community linked to various phases of oral cancer and post-treatment conditions.
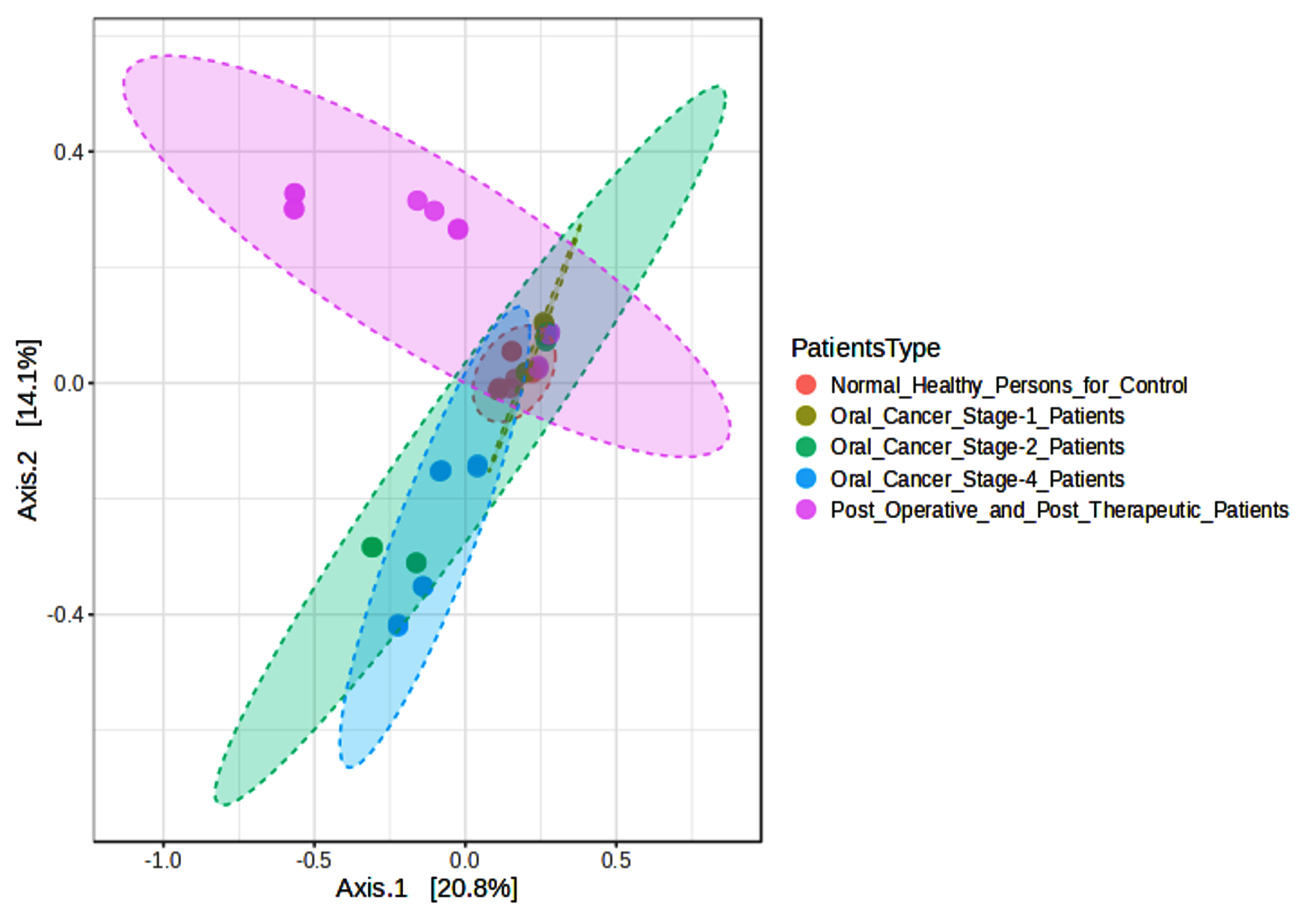
Figure 3. Beta diversity index PCoA Bray-Curtis among groups
44 significant microbial species discriminating between healthy persons, oral cancer patients, and post-operative/post-therapeutic patients were found using an LEfSe analysis with a p-value of 0.01, an FDR adjusted threshold, and an LDA score of 2.0 (Figure 4). The important microbial species that have been found are crucial to the development of oral cancer because they affect immune regulation, tumor microenvironment modifications, and dysbiosis. Prior research has connected Selenomonas timonae and Campylobacter rectus to immunological dysregulation, chronic inflammation, and periodontal disease, all of which are risk factors for cancer development. Similarly, it is known that some species, such as Treponema socranskii and Porphyromonas endodontalis, release virulence factors that encourage inflammatory reactions and tissue destruction, which may facilitate the development of oral cancer. Prevotella species enrichment in cancer patients points to a change in the oral microbiome toward increased inflammation. Patients who had received therapy demonstrated some degree of microbial recovery; species usually linked to oral health and homeostasis, Lactobacillus gasseri and Veillonella dispar, were found to be highly enriched. But the survival of harmful species like Fusobacterium vincentii and Selenomonas sputigena in patients who have finished treatment suggests that full microbial restoration may not happen after therapy, which might lead to a risk of secondary problems or cancer recurrence.
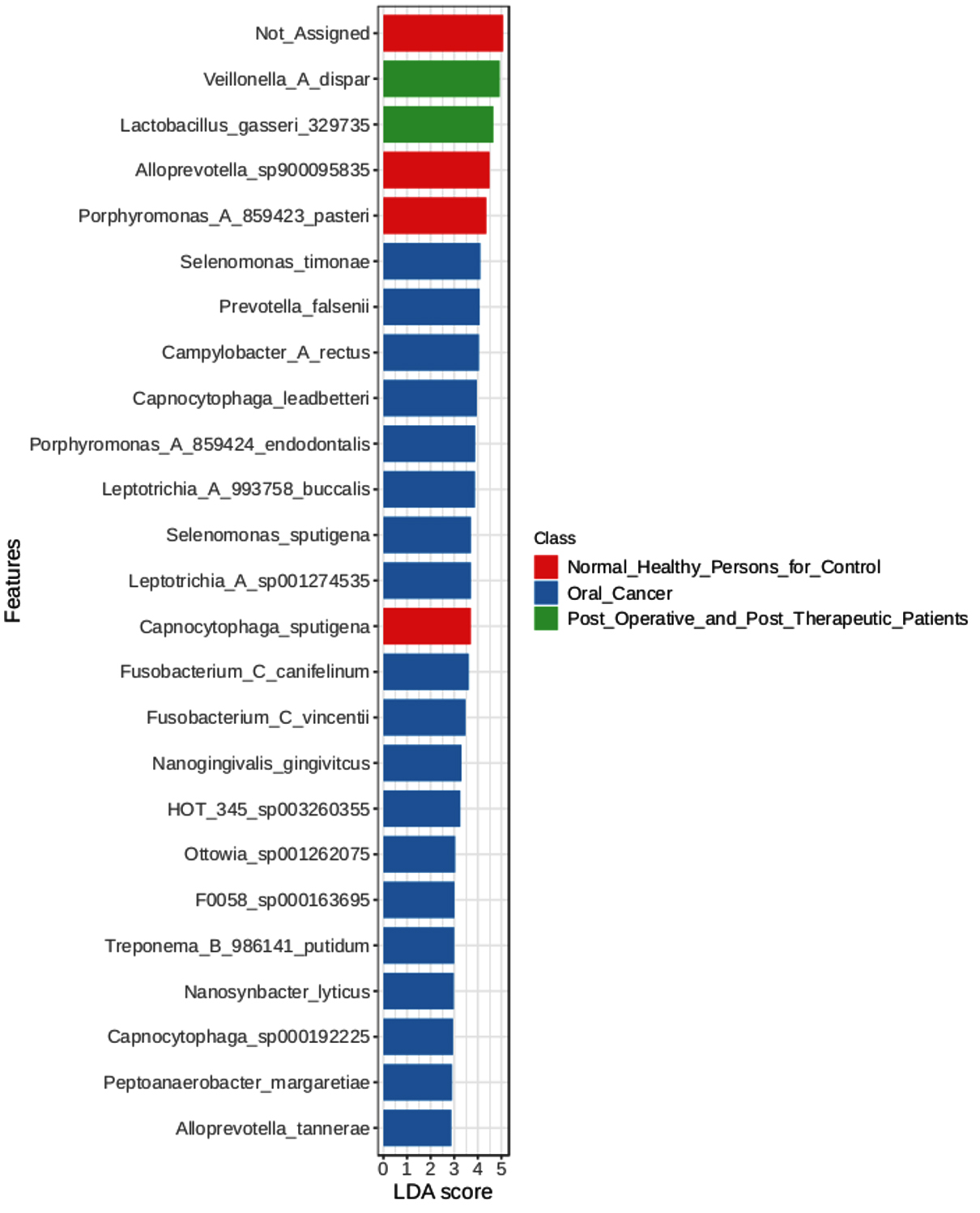
Figure 4. LEfSe Bar Graph
These results emphasize the potential of microbial indicators like Selenomonas timonae and Campylobacter rectus as therapeutic or diagnostic targets and highlight the involvement of the oral microbiome in the advancement of oral cancer. Furthermore, changes in the microbial makeup that have been seen after medication imply that tracking the oral microbiome after treatment may be a useful way to gauge both recovery and long-term health consequences.
LEfSe analysis was performed to identify microbial features with significant differences between buccal and gum samples. This method, which combines statistical significance with biological relevance, highlighted one key microbial features with notable differences: With a linear discriminant analysis (LDA) score of 3.34, the abundance of Rothia mucilaginosa was significantly greater in buccal samples (7094.7) compared to gum samples (2692.3). This suggests that Rothia mucilaginosa may be involved in the different microbial makeup of the buccal cavity as opposed to the gums. It also shows a significant link between the bacteria and the buccal environment. Fluctuations in the local oral environment, such as fluctuations in moisture, pH, or exposure to various food types and microorganisms, may be the cause of these discrepancies (Figure 4).
Multifactor analysis
Our multifactor analysis revealed substantial changes in microbial characteristics across different patient groups across oral cancer stages and post-treatment circumstances in the microbial community. The results indicate that changes in the makeup of the microbial community are linked to the advancement of oral cancer, as demonstrated by the greater number of noteworthy characteristics in Stage 4 of the disease as opposed to Stage 1 or Stage 2. When compared to healthy controls, the post-treatment group showed a comparable number of significant characteristics (27), indicating a partial restoration of microbial diversity and structure after therapy. Nonetheless, the large number of noteworthy characteristics seen between Stage 4 and after treatment (69) suggests that advanced cancer may result in more severe and long-lasting changes in the microbiome that are not entirely restored by therapy. The dynamic variations in microbial profiles that occur as the illness develops are highlighted by the comparison of Stage 1 and Stage 4 (45 important characteristics), suggesting microbial biomarkers that might distinguish between early and late stages of oral cancer. According to the multifactor analysis, the abundance of Desulfobulbus oralis was much higher at oral cancer stage 4 (Figure 5), although it was barely noticeable in earlier stages of the disease, after therapy, and in healthy persons. The sulfate-reducing bacteria Desulfobulbus oralis is well-known for its capacity to generate the genotoxic chemical hydrogen sulfide (H2S). Increased H2S levels have been linked to tumor-promoting microenvironment formation, inflammation, and DNA damage. Particularly in the more advanced phases of the disease, this bacteria may worsen tissue damage and inflammation in the context of oral cancer, creating circumstances that facilitate tumor development and invasion. Given D. oralis’s noteworthy presence in stage 4 cancer, it is possible that the inflammatory reactions and metabolic processes it triggers will be crucial in advancing the disease’s late stages. Furthermore, as the illness progressed from early to late stages, a steady rise in the abundance of Nanosynbacter lyticus was seen (Figure 6), suggesting a potential correlation with the development of cancer. While the precise function of N. lyticus in cancer remains unclear, an increase in its abundance might suggest a dysbiosis that facilitates microbial interactions that advance tumor growth. Such species coexisting with diseases may further upset the delicate microbial equilibrium. These results highlight how crucial it is to monitor microbiological changes along the course of cancer and its recovery in order to have a better understanding of the disease’s related alterations and the effects of treatment therapies.
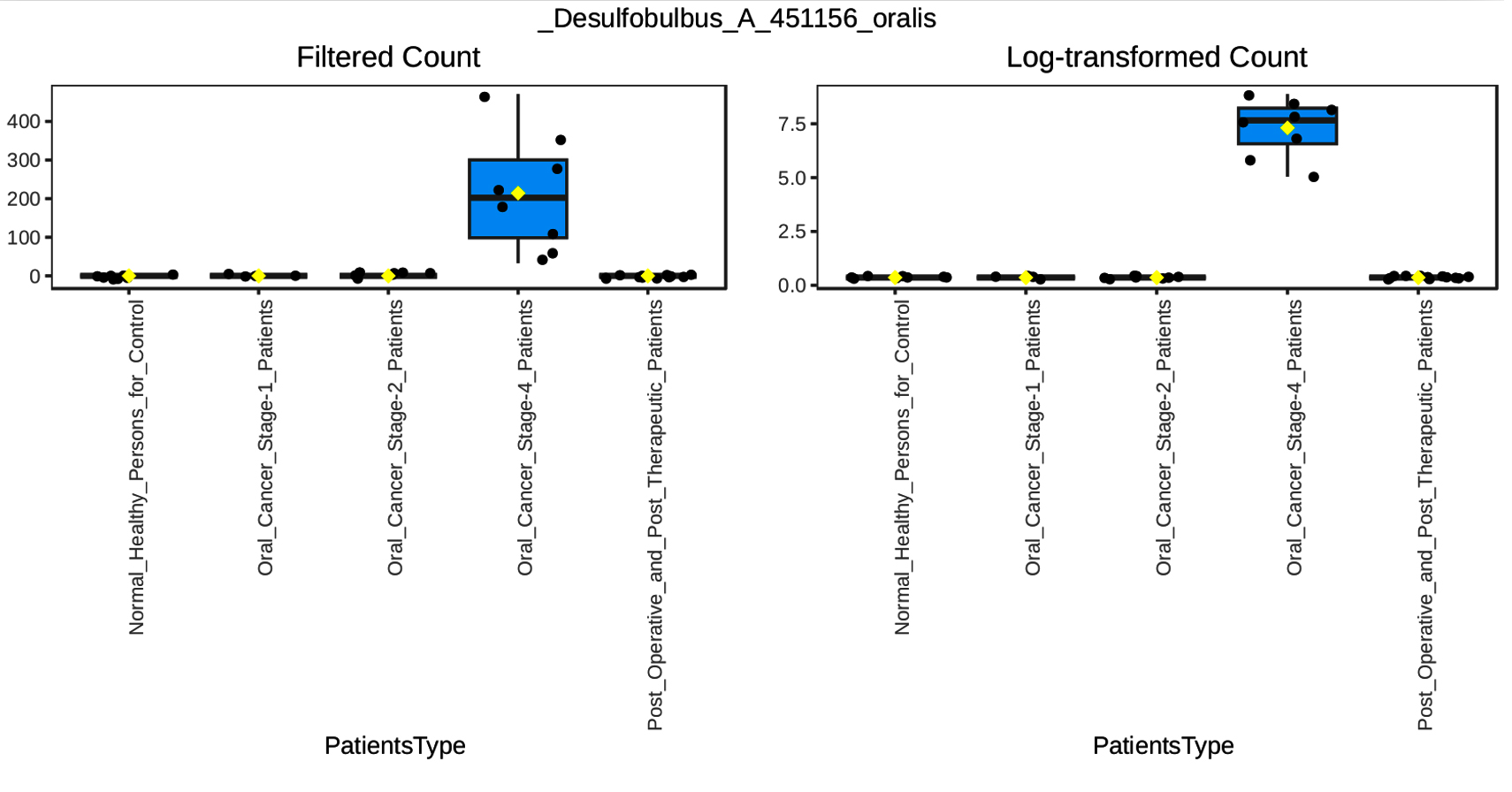
Figure 5. Multifactor Analysis of Desulfobulbus_A_451156_oralis
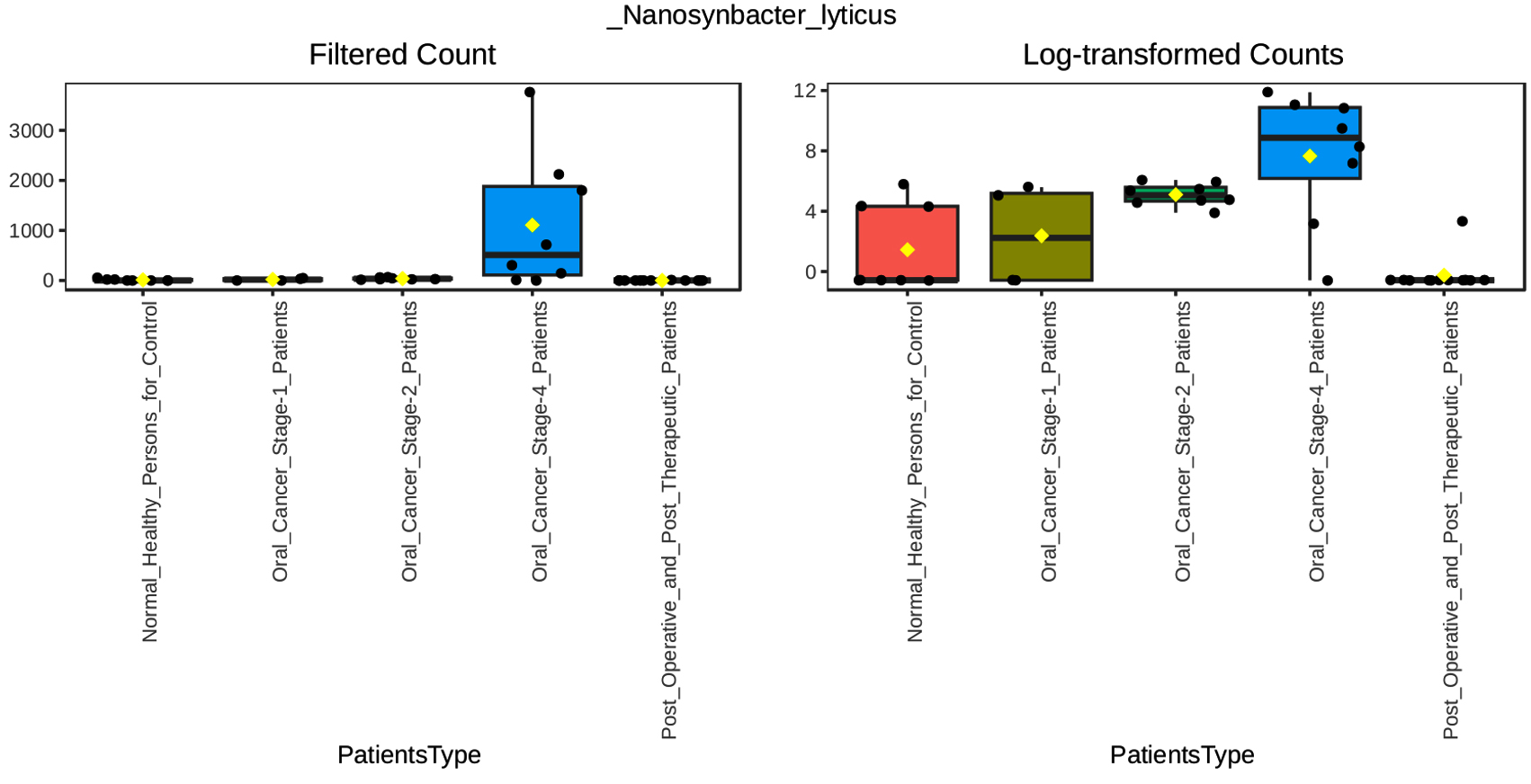
Figure 6. Multifactor Analysis of Nanosynbacter_lyticus
A variety of significant relationships, both positive and negative, between different taxa was shown by the Pearson correlation analysis of microbial taxa in the healthy and stage 4 cancer groups (Figure 7). Among the noteworthy discoveries are:
Strong Positive Correlations
- Prevotella_aurantiaca and Treponema_B_986142_medium (0.8646)
- Veillonella_A_denticariosi and Leptotrichia_A_993758_trevisanii (0.8959)
- Actinomyces_oris_A_386611 and Treponema_B_986142_medium (0.8646)
- Prevotella_seregens and Treponema_B_986142_medium (0.8363)
Strong Negative Correlations
- Alloprevotella_tannerae and Veillonella_A_denticariosi (-0.8031)
- Prevotella_maculosa and Fusobacterium_C_canifelinum (-0.6796)
- Veillonella_A_denticariosi and Nanogingivalis_gingivitcus (-0.7502)
- Veillonella_A_denticariosi and Prevotella_maculosa (-0.8082).
A total of 298 species were identified, with 35 species showing a significantly higher prevalence among those diagnosed with oral cancer compared to those without the disease. A comprehensive analysis revealed that 35 species were absent in persons without any health conditions. Additionally, 12 species were absent in both healthy individuals and patients undergoing post-therapeutic interventions. However, a significantly higher number of species were discovered in patients with oral cancer.
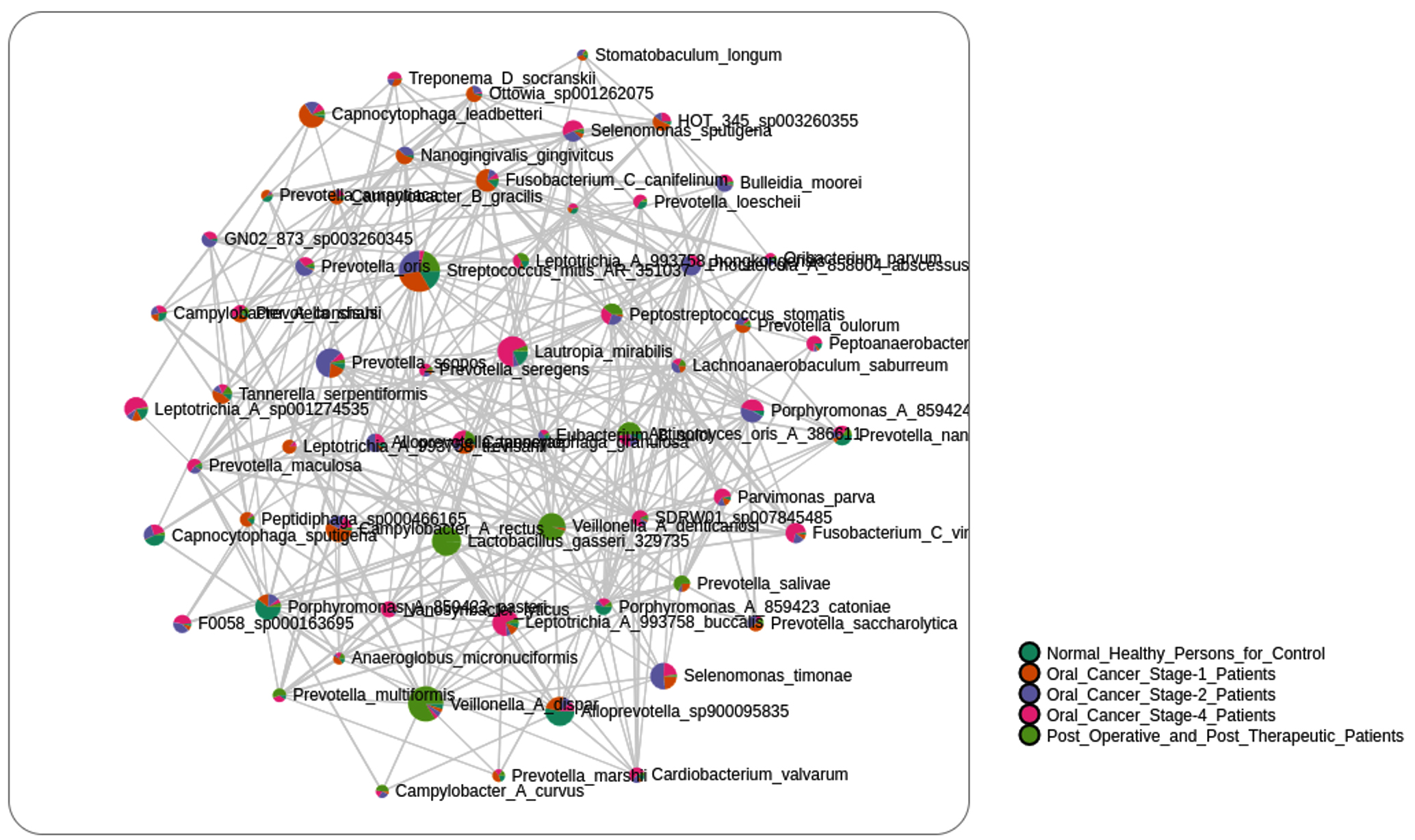
Figure 7. Correlation of Bacterial Species
Acetobacteroides hydrogenigenes were identified in oral cancer patients (0.0000069%). According to research by Su et. al., Acetobacteroides hydrogenigenes is an anaerobic hydrogen-producing bacteria.32 Actinomyces dentalis was originally identified in human dental abscesses by Hall and colleagues in 2005 and in our study it was detected in oral cancer patients (0.000427%), post-treatment patients (0.000106%), and was absent in healthy individuals (0.0%).33 Bilophila wadsworthia was characterized by Burrichter et al.34 In our study, Bilophila wadsworthia, an anaerobic, sulfite-reducing bacterium commonly found in the human gut microbiota, was observed at 0.0000482% in oral cancer patients, but was not detected in post-treatment or healthy individuals.34 Campylobacter curvus has been associated with extraoral abscesses, including within the oral mucosa, as reported by Han et al,35 present study showed its prevalence at 0.000325% in oral cancer patients and 0.000857% in post-treatment patients, while it was absent in healthy controls.35 Ciantar et al.,36 identified Capnocytophaga haemolytica, a novel species found in subgingival plaque, and in present study, this bacterium was only present in oral cancer patients (0.0001722%), with no detection in healthy individuals or post-treatment patients.36 Cryptobacteroides pullicola was originally identified in the chicken gut microbiome through metagenomics and culture techniques, was described by Gilroy et al.,37 this species also found at a prevalence of 0.0001579% in oral cancer patients, while it was absent in both post-treatment and healthy groups.37 Desulfomicrobium orale , an oral sulfate-reducing bacterium implicated in periodontal disease according to Langendijk, was detected at 0.0000041% in oral cancer patients but not in post-treatment or healthy individuals in this study.38 Eubacterium saphenum, a Gram-positive anaerobic bacillus rarely found in healthy oral sites, as reported by Cheeaeman et al.39 which is commonly associated with periodontal disease and other oral infections, was found at a prevalence of 0.0000152% in oral cancer patients, with no detection in healthy or post-treatment groups.39 Faucicola mancuniensis was isolated from the human oropharynx by Humphreys et al.,40 which was present at 0.000352% in oral cancer patients, 0.000001% in healthy volunteers, and 0.0034268% in post-treatment patients in present study.40 Lancefieldella Spp. was found in large numbers in the human oral cavity, as described by Liu et al,41 and was also detected in tobacco chewers and smokers according to Savalia et al,19 and is present study is is highly abundant in oral cancer patients (0.000287%) but absent in healthy individuals.19,41 Leptotrichia A 993758 shahii identified at a prevalence of 0.0014% in both oral cancer and post-treatment patients, was absent in healthy controls. The detected clusters exhibited a notable co-occurrence of Leptotrichia sp., similar to the patterns identified in colorectal malignancies as reported by Amer et al.42 Carlier et al43 found that Moryella indoligenes an anaerobic bacterium isolated from clinical specimens in 2007 and we highly found in oral cancer patients (0.0005569%), and not found in normal persons and post therapeutic patients.43 In our research, oral cancer had 0.0002587%, and normal people and post therapeutic people had 0.0% of Prevotella buccalis. Prevotella falsenii and Prevotella seregens this both species was found in tobacco counsumers in previous study by Savalia et al,19 in presnt study we identified that oral cancer had high prevelance of Prevotella falsenii (0.01887%) and Prevotella seregens (0.00060%) with campare to normal persons and post therapeutics patients.19 Pseudoramibacter alactolyticus was previously found in primary endodontic infected patients who had been sent for root canal therapy by siquaira et al44 it was present in oral cancer patients (0.0000044%) but absent in normal and post therapeutic patients.44 As per our study prevalence of 0.0000206% of Treponema D parvum was observed in individuals with oral cancer, but it was not found in normal patients or those who had undergone therapy. The previous study conducted by Wyss et al.45 involved the isolation of a novel acid-dependent oral spirochaete species from subjects with human periodontitis and acute necrotizing ulcerative gingivitis.45
Centipeda infelix, Ezakiella massiliensis, Gemella bergeri, Marinomonas communis, Mobiluncus mulieris, Pauljensenia georgiae, Thalassotalea C fusca, and Zavarzinia compransoris this 8 species are present in oral cancer patients but absent in normal healthy individuals as well as also absent in post-therapeutic patients and there is no evidence or previous research regarding these species in the oral area of those patients who suffer from oral cavity and oral cancer.25
Oral cancer and post-treatment microbiomes: A KEGG pathway analysis
Additionally, the Tax4Fun2 package in R was used to carry out functional predictions based on 16S rRNA gene data. The functional profiles of individuals with oral cancer, those in good health, and those who had finished treatment were mapped onto cancer-related pathways using the KEGG Orthology (KO) database. Across the three settings, a number of important metabolic and signaling pathways linked to carcinogenesis, cancer development, and post-treatment changes were found. Significant alterations in the expression of important proteins and enzymes involved in choline metabolism, chemical carcinogenesis, proteoglycan synthesis, central carbon metabolism, and reactive oxygen species (ROS) production were found by the investigation.
Cancer’s central carbon metabolism (ko05230)
When compared to healthy persons, oral cancer patients had a considerable overexpression of the central carbon metabolism pathway, especially for enzymes like pyruvate dehydrogenase (PDHA and PDHB) and L-lactate dehydrogenase (LDH). These enzymes play a critical role in the Warburg effect, a glycolytic shift frequently seen in cancer cells. In comparison to controls, the counts of these enzymes were higher in cancer patients, suggesting that during carcinogenesis, there was an increase in glycolysis and a change in energy metabolism. Samples taken after treatment showed a partial reversal of this upregulation, although LDH levels remained elevated, indicating that metabolic changes persisted even after treatment.
Cancer pathways (ko05200)
Patients with cancer had higher levels of many cancer pathways, such as glutathione S-transferase (GST) and NAD(P)H dehydrogenase (NQO1), especially at Stage 4, suggesting greater oxidative stress and detoxification mechanisms in advanced disease. This held true for both buccal and gum swabs, the two body locations. While levels remained increased relative to healthy controls, post-treatment patients showed decreased counts for these enzymes, suggesting reduced stress following therapy, indicating continued adaptation to oxidative damage or medication-induced stress.
ROS generation and chemical carcinogenesis (ko05208)
Patients with oral cancer showed significant elevation of the reactive oxygen species (ROS) pathway (ko05208), especially for enzymes such as catalase (katE) and superoxide dismutase (SOD1, SOD2). In order to neutralize ROS produced during carcinogenesis, these enzymes are essential. According to the count data, ROS-scavenging enzyme levels were considerably greater in cancer patients, particularly in those in Stage 4. This was probably a defensive reaction to increased oxidative stress. Samples taken after therapy revealed a drop in ROS-related enzymes, but these levels did not increase back to normal, suggesting persistent oxidative stress.
Cancer-related choline metabolism (ko05231)
Patients with cancer had significant changes in the choline metabolism pathway, including increased activity of enzymes including phospholipase D1/2 (PLD1/2) and diacylglycerol kinase (dgkA). These enzymes have a role in membrane formation and lipid signaling, two processes that are often dysregulated in cancer. These enzymes showed partial downregulation in post-treatment samples, indicating a partial recovery of normal lipid metabolism following medication.
This work highlights important microbial and functional alterations related with the progression and recovery from oral cancer. It offers thorough insights into the microbiome dynamics across healthy, oral cancer, and post-treatment patient groups. Important discoveries from diversity indices and LEfSe investigations show that, in comparison to healthy persons, oral cancer stages, particularly Stage 4, are characterized by a unique microbial shift, with species including Campylobacter rectus and Selenomonas timonae associated with tumor-promoting conditions. Additionally, partial microbial diversity restoration was observed in post-treatment samples; nevertheless, certain pathogenic species, such as Fusobacterium vincentii, continued to exist. These findings raise the possibility that full microbial restoration may not occur post-treatment, which might increase the risk of cancer recurrence. Significant changes were found in cancer-related metabolic pathways, namely in the areas of central carbon metabolism, ROS generation, and chemical carcinogenesis pathways, according to functional pathway analysis using KEGG. These modifications show elevated oxidative stress and glycolysis in cancer patients, with a partial recovery shown after treatment; nevertheless, certain metabolic disturbances continued, indicating the long-term effects of the illness and treatment. The study emphasizes how crucial it is to keep an eye on functional and microbiological changes in patients with oral cancer in order to better understand the course of the illness, possible targets for treatment, and long-term results. The study’s discovery of microbial biomarkers, especially those connected to dysbiosis and altered tumor microenvironment, may be used to inform prognostic, therapeutic, and diagnostic approaches for the treatment of oral cancer.
ACKNOWLEDGMENTS
The authors express their heartfelt gratitude to Sterling Accuris Diagnostics-Ahmedabad and the Gujarat Biotechnology Research Centre (GBRC), Gandhinagar, for their indispensable assistance in supplying the necessary laboratory resources that successfully enabled this research. Furthermore, we would like to extend our appreciation to the study participants and Sterling Hospitals, Ahmedabad, for authorising the collection of the necessary samples for this research.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
None.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript.
ETHICS STATEMENT
This study was approved by the Sterling Hospital Ethics Committee, Ahmedabad, Gujarat, India (SHEC/HS/OC-Study/235-2022).
INFORMED CONSENT
Written informed consent was obtained from the participants before enrolling in the study.
- Abati S, Bramati C, Bondi S, Lissoni A, Trimarchi M. Oral cancer and precancer: A narrative review on the relevance of early diagnosis. Int J Environ Res Public Health. 2020;17(24):1-14.
Crossref - Ren ZH, Hu CY, He HR, Li YJ, Lyu J. Global and regional burdens of oral cancer from 1990 to 2017: Results from the global burden of disease study. Cancer Commun. 2020;40(2-3):81-92.
Crossref - Borse V, Konwar AN, Buragohain P. Oral cancer diagnosis and perspectives in India. Sensors Int. 2020;1:100046.
Crossref - Kimple AJ, Welch CM, Zevallos JP, Patel SN. Oral Cavity Squamous Cell Carcinoma – An Overview. Oral Health Dent Manag. 2014;13(3):877-882.
- Wong TSC, Wiesenfeld D. Oral Cancer. Aust Dent J. 2018;63(Suppl 1):S91-S99.
Crossref - Varshitha A. Prevalence of Oral Cancer in India. J Pharm Sci Res. 2015;7(10):845-848.
- Dhanuthai K, Rojanawatsirivej S, Thosaporn W, et al. Oral cancer: A multicenter study. Med Oral Patol Oral Cir Bucal. 2018;23(1):e23-e29.
Crossref - Tuominen H, Rautava J. Oral Microbiota and Cancer Development. Pathobiology. 2021;88(2):116-126.
Crossref - Lopez-Jornet P, Hynninen JN, Parra-Perez F, Peres-Rubio C, Pons-Fuster E, Tvarijonaviciute A. The Role of Salivary Biomarkers in Monitoring Oral Health in Patients with Implants and Periodontitis. Appl Sci. 2024;14(2):927.
Crossref - Avila M, Ojcius DM, Zlem Yilmaz O. The Oral Microbiota: Living with a Permanent Guest. DNA Cell Biol. 2009;28(8):405-411.
Crossref - Chattopadhyay I, Verma M, Panda M. Role of Oral Microbiome Signatures in Diagnosis and Prognosis of Oral Cancer. Technol Cancer Res Treat. 2019;18:1533033819867354.
Crossref - Sender R, Fuchs S, Milo R. Revised Estimates for the Number of Human and Bacteria Cells in the Body. PLoS Biol. 2016;14(8):1-14.
Crossref - Vakilzadeh MM, Khayami R, Daneshdoust D, et al. Prevalence of tobacco use among cancer patients in Iran: a systematic review and meta-analysis. BMC Public Health. 2024;24(1):1081.
Crossref - Homann N, Tillonen J, Rintamaki H, Salaspuro M, Lindqvist C, Meurman JH. Poor dental status increases acetaldehyde production from ethanol in saliva: A possible link to increased oral cancer risk among heavy drinkers. Oral Oncol. 2001;37(2):153-158.
Crossref - Mohammed RA, Ahmed SK. Oral cancer screening: Past, present, and future perspectives. Oral Oncol Reports. 2024;10:100306.
Crossref - Khan MF, Hayhoe RP, Kabir R. Exploring the Risk Factors for Oral Cancer in Pakistan: A Systematic Literature Review. Dent J. 2024;12(2):25.
Crossref - Perera M, Al-Hebshi NN, Speicher DJ, Perera I, Johnson NW. Emerging role of bacteria in oral carcinogenesis: A review with special reference to perio-pathogenic bacteria. J Oral Microbiol. 2016;8(1):32762.
Crossref - Wirth R, Maroti G, Mihok R, et al. A case study of salivary microbiome in smokers and non-smokers in Hungary: analysis by shotgun metagenome sequencing. J Oral Microbiol. 2020;12(1):1773067.
Crossref - Savalia HJ, Patel N, Singh KM, et al. Dominance of Prevotella Species in Tobacco Consumers: A Metagenomic Preliminary Study. Biosci Biotechnol Res Asia. 2024;21(2):671-687.
Crossref - Banerjee J, Mishra N, Dhas Y. Metagenomics: A new horizon in cancer research. Meta Gene. 2015;5:84-89.
Crossref - Handelsman J, Tiedje J, Alvarez-Cohen L, et al. The New Science of Metagenomics: Revealing the Secrets of Our Microbial Planet. The National Academies Press, Washington DC, 2007.
Crossref - Li Q, Hu Y, Zhou X, Liu S, Han Q, Cheng L. Role of oral bacteria in the development of oral squamous cell carcinoma. Cancers. 2020;12(10):1-18.
Crossref - Chiu CY, Miller SA. Clinical metagenomics. Nat Rev Genet. 2019;20(6):341-355.
Crossref - Xu P, Gunsolley J. Application of metagenomics in understanding oral health and disease. Virulence. 2014;5(3):424-432.
Crossref - Illumina. 16S Metagenomic Sequencing Library Preparation. Preparing 16S Ribosomal RNA Gene Amplicons for the Illumina MiSeq System. 2013;(B):1-28. http://support.illumina.com/content/dam/illumina-support/documents/documentation/chemistry_documentation/16s/16s-metagenomic-library-prep-guide-15044223-b.pdf
- Cantu VA, Sadural J, Edwards R. PRINSEQ++, a multi-threaded tool for fast and efficient quality control and preprocessing of sequencing datasets. PeerJ Prepr. 2019;7(2019):e27553v1.
Crossref - Bolyen E, Rideout JR, Dillon MR, et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat Biotechnol. 2019;37(8):852-857.
Crossref - Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP. DADA2: High-resolution sample inference from Illumina amplicon data. Nat Methods. 2016;13(7):581-583.
Crossref - Dhariwal A, Chong J, Habib S, King IL, Agellon LB, Xia J. Microbiome Analyst: A web-based tool for comprehensive statistical, visual and meta-analysis of microbiome data. Nucleic Acids Res. 2017;45(W1):W180-W188.
Crossref - Chong J, Liu P, Zhou G, Xia J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat Protoc. 2020;15(3):799-821.
Crossref - Lu Y, Zhou G, Ewald J, Pang Z, Shiri T, Xia J. Microbiome Analyst 2.0: Comprehensive statistical, functional and integrative analysis of microbiome data. Nucleic Acids Res. 2023;51(W1):W310-W318.
Crossref - Su XL, Tian Q, Zhang J, et al. Acetobacteroides hydrogenigenes gen. nov., Sp. nov., An anaerobic hydrogen-producing bacterium in the family Rikenellaceae isolated from a reed swamp. Int J Syst Evol Microbiol. 2014;64(Pt 9):2986-2991.
Crossref - Hall V, Collins MD, Lawson PA, Falsen E, Duerden BI. Actinomyces dentalis sp. nov., from a human dental abscess. Int J Syst Evol Microbiol. 2005;55(1):427-431.
Crossref - Burrichter AG, Dorr S, Bergmann P, et al. Bacterial microcompartments for isethionate desulfonation in the taurine-degrading human-gut bacterium Bilophila wadsworthia. BMC Microbiol. 2021;21(1):1-16.
Crossref - Han XY, Tarrand JJ, Rice DC. Oral Campylobacter species involved in extraoral abscess: A report of three cases. J Clin Microbiol. 2005;43(5):2513-2515.
Crossref - Ciantar M, Spratt DA, Newman HN, Wilson M. Capnocytophaga granulosa and Capnocytophaga haemolytica: Novel species in subgingival plaque. J Clin Periodontol. 2001;28(7):701-705.
Crossref - Gilroy R, Ravi A, Getino M, et al. Extensive microbial diversity within the chicken gut microbiome revealed by metagenomics and culture. Peer J. 2021;9:1-142.
Crossref - Langendijk PS, Kulik EM, Sandmeier H, Meyer J, van der Hoeven JS. Isolation of Desulfomicrobium orale sp. nov. and Desulfovibrio strain NY682, oral sulfate-reducing bacteria involved in human periodontal disease. Int J Syst Evol Microbiol. 2001;51(3):1035-1044.
Crossref - Cheeaeman SL, Hiom SJ, Weightman AJ, Wade WG. Phylogeny of Oral Asaccharolytic Eubacterium Species Determined by 16S Ribosomal DNA Sequence Comparison and Proposal of Eubacterium infirmum sp. nov. and Eubacterium tardum sp. nov. Int J Syst Bacteriol. 1996;46(4):957-959.
Crossref - Humphreys GJ, Oates A, Ledder RG, McBain AJ. Faucicola mancuniensis gen. nov., sp. nov., isolated from the human oropharynx. Int J Syst Evol Microbiol. 2015;65(1):11-14.
Crossref - Liu G, Tang CM, Exley RM. Non-pathogenic Neisseria: Members of an abundant, multi-habitat, diverse genus. Microbiol (United Kingdom). 2015;161(7):1297-1312.
Crossref - Amer A, Galvin S, Healy CM, Moran GP. The microbiome of potentially malignant oral leukoplakia exhibits enrichment for Fusobacterium, Leptotrichia, Campylobacter, and Rothia species. Front Microbiol. 2017;8(DEC):1-9.
Crossref - Carlier JP, K’ouas G, Han XY. Moryella indoligenes gen. nov., sp. nov., an anaerobic bacterium isolated from clinical specimens. Int J Syst Evol Microbiol. 2007;57(4):725-729.
Crossref - Siqueira JF, Rocas IN. Pseudoramibacter alactolyticus in primary endodontic infections. J Endod. 2003;29(11):735-738.
Crossref - Wyss C, Dewhirst FE, Gmur R, et al. Treponema parvum sp. nov., a small, glucuronic or galacturonic acid-dependent oral spirochaete from lesions of human periodontitis and acute necrotizing ulcerative gingivitis. Int J Syst Evol Microbiol. 2001;51(3):955-962.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.