


ISSN: 0973-7510
E-ISSN: 2581-690X
The convergence of HIV-1, Mycobacterium tuberculosis, and SARS-CoV-2 infections presents a formidable challenge characterized by heightened immune suppression and persistent viral replication. Understanding the shared molecular mechanisms underlying these co-infections is essential, providing a foundation for future translational research and therapeutic discovery. We analyzed differentially expressed genes from HIV-1/TB co-infection datasets across multiple tissues, including lung, spleen, liver, and whole blood. This was followed by PPI network analysis and single-cell multi-gene expression analysis. Gene set enrichment and pathways enrichment analysis were carried out to reveal the immune regulatory pathways that are altered during HIV-1/TB infection and SARS-CoV-2 infection. Gene expression profiling pointed out critical genes, including CXCR4, TAP1, TSC22D3 and FGR, associated with immune suppression, antigen processing, and viral attachment. TSC22D3 and CXCR4 were found to be crucial immune cell regulators in lung tissue using single-cell analysis. Apart from this, PPI network analysis revealed that TAP1, TAP2, and CXCR4 are essential hub genes, whereas the intersectional analysis with the SARS-CoV-2 gene datasets highlighted 27 genes associated with the immune response and viral persistence. Pathway enrichment analyses noted several processes, such as protein phosphorylation, apoptosis, and immune evasion, pointing out common mechanisms across these infections. Our findings suggest shared molecular signatures in HIV-1/TB and SARS-CoV-2 infections, with central genes offering potential therapeutic relevance. Future work will validate these targets through wet lab experiments to better understand their roles in immune regulation and develop novel therapies to enhance viral clearance in co-infected patients.
Mycobacterium tuberculosis, HIV-1, SARS-CoV-2, Gene Expression, Gene Ontology, Functional Analysis, Systems Biology
The co-infection of HIV-1 and Mycobacterium tuberculosis (Mtb) is one of the most significant global public health challenges, particularly in places where both illnesses are widespread. Tuberculosis (TB) is a highly infectious disease caused by Mtb, affecting millions globally.1,2 It is estimated that one-third of the world’s population is infected with Mtb, with 5-10% developing active TB during their lifetime. The primary site of Mtb infection is the lungs, leading to pulmonary TB, which can worsen progressively if not treated. However, TB is not only limited to the lungs; it can invade other organs, such as the urinary tract, lymph nodes, and even the brain, leading to extrapulmonary TB.2,3 Even with the significant advancements made in the treatment of tuberculosis (TB), its impact in terms of morbidity and mortality remains a serious public health problem worldwide. According to the report of the World Health Organization, 13 lakh people died due to tuberculosis in the year 2022.4 Inflammation caused by HIV-1 infection impairs the immune system by lowering CD4+ T cells which can trigger acquired immunodeficiency syndrome (AIDS) without any treatment.5 Having TB and HIV-1 together causes a heightened level of immune deficiency, a more rapid onset of disease, and an increased risk of death. TB incidence among those with HIV is a significant concern, as about 20% of those living with HIV/AIDS will develop active TB.6,7 The function of CD4+ T cells is critical to controlling immune responses to Mtb infections. They also help in the augmentation of CD8+ T cells, the cells containing the cytotoxic activity, and other immune cells that produce effector molecules, such as IFN-gamma, essential in bacterial replication control.8,9 Such CD4+ T cell depletion as a consequence of HIV-1 infection can be detrimental to the self-regulatory aspect of the immune system and accelerate the transition from latent to active. This fall raises the chances of contracting tuberculosis in addition to inhibiting the immune response, and thus, there are higher chances of TB reactivation and mortality. TB and HIV-1 have been shown to operate interactively in a way that explains many aspects of their detrimental concurrency.10,11 The deficiency of CD4+ T cells enhanced by HIV-1 contributes to TB, but TB itself may contribute to HIV-1 spread at the region of TB, such as tuberculous pleural effusions (TPE). TB-PE, a widespread extrapulmonary condition, features the buildup of pleural fluid between the lungs and the chest cavity, increasing immune cells’ vulnerability to HIV-1 infection and boosting the persistence of the virus.11,12 Still, specific evidence suggests that Mtb might inhibit HIV-1 replication in some cases, which stresses the complex nature of these interactions.
Furthermore, the emergence of SARS-CoV-2 complicates things even more. SARS-CoV-2 predominantly impacts the respiratory system but can also affect immune function by initiating cytokine release and lessening T cells.13,14 Previously, it has been shown that SARS-CoV-2 can co-infect those who are HIV-1 or TB-positive, and this association results in worse clinical outcomes from common immune evasion, viral replication, and immune dysfunction. Responses of CD4+ T cells to SARS-CoV-2 are essential for building lasting immunity and subduing infection. People infected with both SARS-CoV-2 and HIV-1 suffer from more significant immune dysfunction caused by CD4+ T cell depletion, which lengthens the duration of their illness and raises their mortality risk.15,16 Understanding typical molecular mechanisms related to these co-infections is of great importance because of the complicated interactions associated with HIV-1, Mtb, and SARS-CoV-2. Though research has affirmed a bidirectional effect between HIV-1 and Mtb in terms of their pathogenesis, there is an ongoing gap in comprehensive knowledge about these pathogens at the molecular level, particularly for co-infection with SARS-CoV-2.15,17 Such immune dysfunction is probably due to the common mechanisms responsible for immunosuppression, viral persistence, and immune escape. Subjects with co-infection have significantly reduced CD4+ T cells, increasing the chances of reactivation and advancing to active disease, in contrast to those with single infections.18 The resulting drop in CD4+ T cell counts by SARS-CoV-2 complicates things by undermining immune responses.
To tackle these challenges, bioinformatics and systems biology afford essential techniques for understanding the molecular interactions involving HIV-1, Mtb, and SARS-CoV-2. We aim to apply large-scale gene expression data, protein-protein interaction networks, and pathway enrichment analyses to identify the common molecular determinants among these pathogens.19 Notably, multi-omics, such as tissue and single-cell-based gene expression profiling, elucidate how the pathogens hijack the host immune system. Targeting critical regulatory centers or pathways may provide us with the potential strategy for the treatment of co-infectious diseases in a more effective manner. In this study, we sought to explore the molecular pathways and immune regulatory mechanisms underlying co-infections with HIV-1, TB, and SARS-CoV-2. Using the above methods, we analyzed the dynamic patterns of gene expression and host-pathogen interactions that impaired immune signals and favored the maintenance of the virus and evasion from immunity. The workflow of the study is given in Figure 1. This comprehensive approach allowed us to identify fundamental regulatory mechanisms and shared molecular pathways, offering new insights that could inform future therapeutic strategies for enhancing immune responses in co-infected individuals (Figure 1).
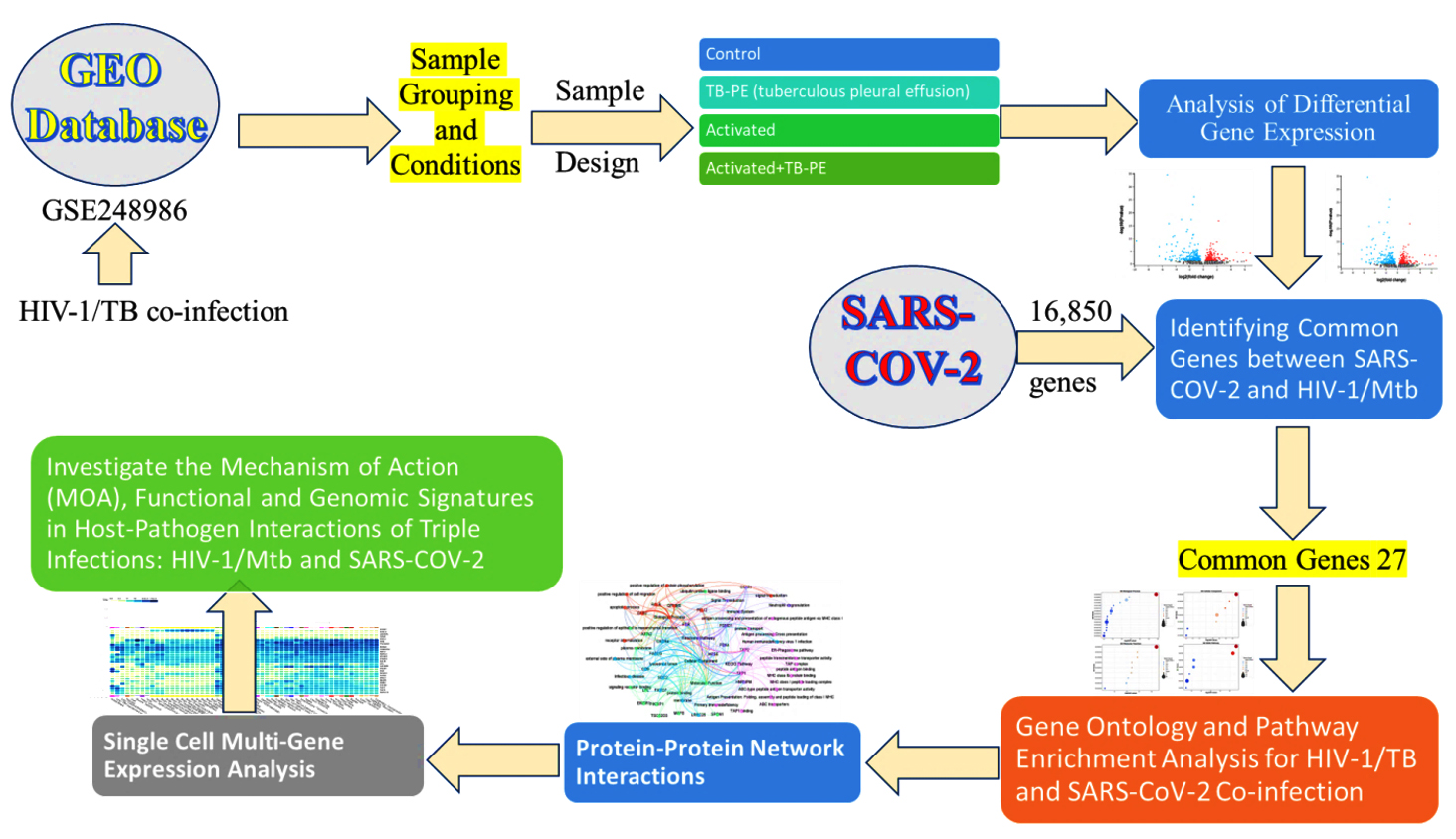
Figure 1. The workflow design of the study illustrates the key steps and methodologies employed in the analysis, from data collection to the results
Dataset acquisition and experimental design
The transcriptomic data relevant to this study was downloaded from the NCBI GEO database,20 and the accession number GSE248986 was used.12 This set includes mRNA expression profiles of CD4+ T cells from three HIV-negative healthy or Mtb-exposed donors (Donors, 30, 31, 32). The study aims to measure how the environmental factors associated with tuberculosis influence the propagation and persistence of HIV-1 in CD4+ T cells.12
Sample grouping and conditions
The sample contains twelve samples from four experimental groups: Control, TB-PE (tuberculous pleural effusion), Activated, and Activated+TB-PE. CD4+ T cells isolated from healthy donors served as the Control group. The TB-PE group consisted of CD4+ T cells treated with the acellular fraction of TB-PE to simulate the tuberculosis-associated microenvironment. The Activated group involved CD4+ T cells activated with anti-CD3/CD28 antibodies to mimic immune activation. The final group, Activated+TB-PE, included CD4+ T cells simultaneously activated with anti-CD3/CD28 and exposed to TB-PE. Each donor provided samples across all four experimental conditions, ensuring consistent group comparisons.
RNA sequencing and data processing
The Illumina NovaSeq 6000 platform performed RNA sequencing on mRNA expression data from 12 samples (GPL24676). Before further analyses, the raw expression data were pre-processed to make them comparable across different sources and eliminate batch and other confounding effects. Data normalization was performed using the Robust Multi-array Average (RMA) method, followed by log2 transformation to reduce variability in the data. Such data normalization allowed unbiased gene expression comparison across various treatments and all other experiment conditions.
Differential gene expression
To identify differentially expressed genes (DEGs), GEO2R was employed to design an R script to analyze the RNA-seq data GSE248986. The R version 4.2.2 and several Bioconductor packages, including GEOquery (2.66.0), limma (3.54.0), and DESeq2, were employed in this process. The GEO-derived raw count data were also internally retrieved along with the pre-processed and normalized RNA-seq data obtained using DESeq2. The DESeq2 algorithm was utilized to carry out the differential expression analysis. At first, low-count genes were excluded, retaining genes with at least ten counts across samples. The expression of genes was normalized using the DESeq2 method, comprising a multi-factor analysis registered to tick the presence of DEGs for all the contrast comparisons. Following the aforementioned stepwise examination of over-expressed DEGs, the Wald test was implemented to examine the in-between differences between the clinical groups. For the results, the adjustment was made for multiplicity using the Benjamini-Hochberg FDR. DEGs with an adjusted p-value <0.05 and absolute log₂ fold change ≥1 were considered statistically significant. In addition, volcano plots were used to show the DEG distribution, and a Venn diagram was utilized to assess the number of those genes shared by DEGs for various comparisons.
Curating genes from the CTD database and identifying common genes
After identifying the 31 common genes from the Venn diagram analysis using GEO2R from five distinct groups, these genes were then cross-referenced with genes related to SARS-CoV-2 from the Comparative Toxicogenomics Database (CTD).21 This step aimed to determine which shared genes from the Tuberculosis-associated microenvironment study also directed activities in the SARS-CoV-2 pathways. Such genes that have significant roles in both conditions were highlighted, which would mean that such diseases have a certain degree of similar genetic regulatory mechanisms. This approach helped us narrow the focus and analyze the genes involved in the pathophysiological overlap of tuberculosis, HIV-1 latency, and COVID-19 and strategize forward.
Gene ontology and pathway enrichment analysis for HIV-1/TB and SARS-CoV-2 co-infection
The Genes common from the analysis of co-infections of HIV/TB with SARS-CoV-2 were then analyzed for GO and Pathway Enrichment Analysis for biological purposes. Data analysis was done through the DAVID tool to assess biological processes, cellular components, and molecular functions of genes. The enrichment analysis was supplemented with the KEGG22 and Reactome pathways databases to elucidate comprehensively the signaling pathways and molecular interactions these genes affect. Additionally, GSEA was performed using the Enricher platform,23 incorporating multiple gene set libraries such as ChEA 2022, KEGG 2021 Human, and the Elsevier Pathway Collection. This allowed for a more comprehensive investigation into the shared pathways and molecular interactions that drive the immune dysregulation and viral persistence observed in these co-infections.
Protein-protein network tnteractions
To analyze the functional interactions of genes originating from HIV-1/TB and SARS-CoV-2 co-infections, we deployed protein-protein interaction (PPI) network analysis. Protein-protein interaction (PPI) networks were predicted and constructed using the STRING database,24 which used high confidence (≥0.7) to ensure that all valid interactions were captured within the network. The PPI network was visualized and further analyzed using Cytoscape25 and Gephi software.26 The PPI Network analysis showed the presence of crucial hub genes functional in immune responses, controlling the viral load, and controlling immune response and evasion by calculating PPI network graphical parameters like degree, betweenness centrality, and closeness centrality. Furthermore, modularity analysis was also done to identify functional groups of the network related to the presentation of antigens, signaling and entry of viruses.
Single cell multi-gene expression analysis in lung tissue
Based on the results obtained from the Gene Ontology and Pathway Enrichment Analysis, GTEx Single Cell Multi-Gene Expression Analysis27 was conducted to examine the expression profiles of the commonly surveyed genes at the lung tissue level. This focus was particularly relevant, as the lungs are a primary site of infection for both SARS-CoV-2 and Mycobacterium tuberculosis, making them a critical tissue for understanding the underlying biology of viral propagation, immune evasion, and co-infection strategies in specific lung cell types.
Differential gene expression analysis
The analysis of differentially expressed genes (DEGs) in the HIV-1/TB co-infection dataset (GSE248986) aimed to enhance our understanding of the mechanisms underlying tuberculosis pleural effusion (TB-PE), HIV-1 latency, and T cell activation. These observations corresponded to a systematic and the highest performance correlation in the profiles for upregulation and downregulation of expression of genes when various conditions of Control, TB-PE, Activated, and Activated_TB-PE were applied and based on a set statistical log2 fold change and Benjamini-Hochberg adjusted p-value (padj <0.05) (Table S1-S5). In the interaction of Control and TB-PE conditions, 433 DEGs have been identified, of which 207 were upregulated and 226 downregulated. This suggests that TB-PE exerts a considerable effect on gene expression, likely contributing to immune suppression and viral persistence. When comparing Control vs Activated, a more significant number of DEGs (8096) were observed, split almost equally between upregulated (4024) and downregulated (4072) genes (Figure 2). This reflects the robust activation of immune responses upon T-cell stimulation.
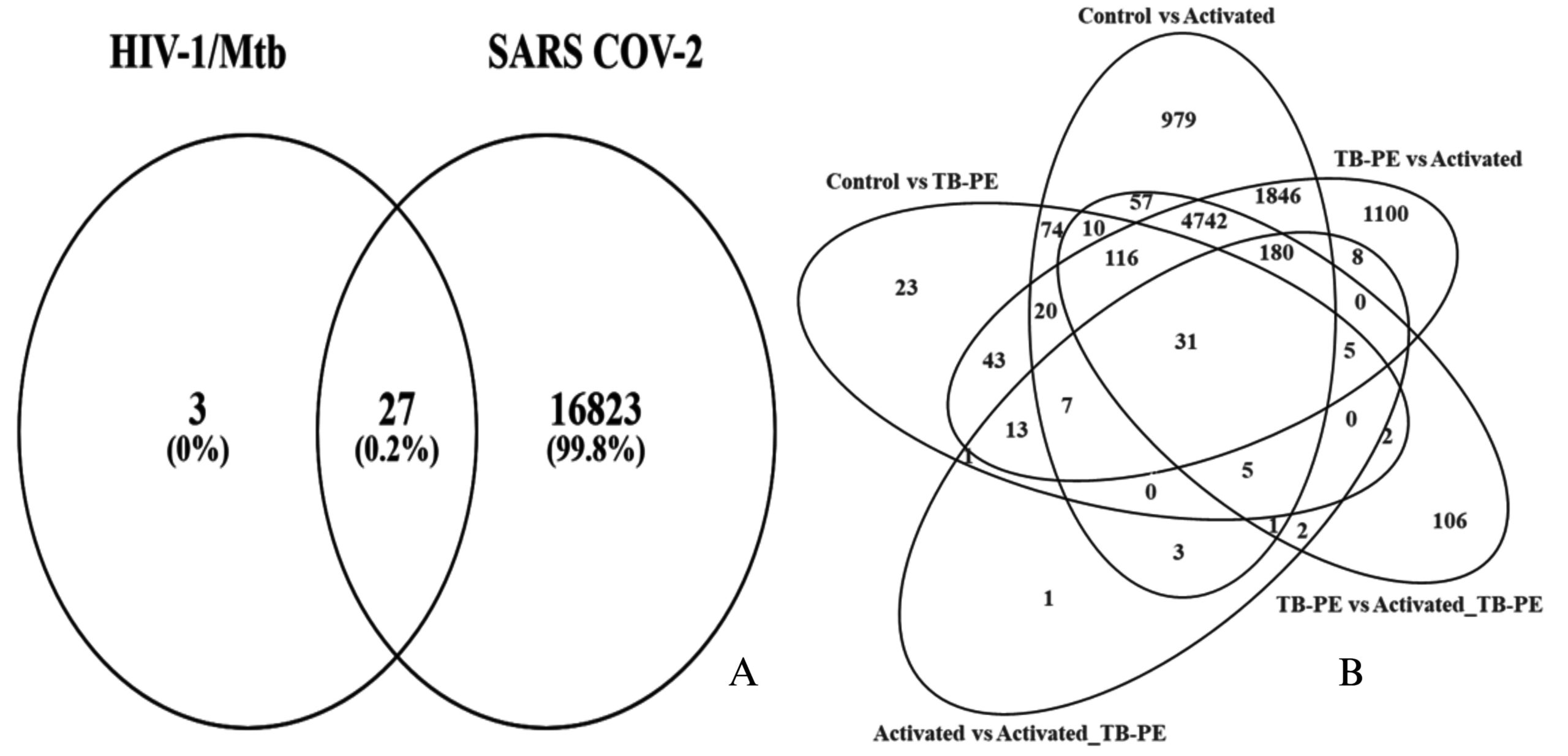
Figure 2. The image displays a Venn diagram; (A) The common genes between identified groups for differentially expressed genes (DEGs); (B) illustrates the common genes between HIV-1/Mtb and SARS-CoV-2
Interestingly, the TB-PE vs Activated comparison showed an even higher number of DEGs (8629), with 4321 genes upregulated and 4308 downregulated, indicating a dynamic shift in gene regulation when immune cells transition from a suppressed state (under TB-PE influence) to an activated state. The comparison between TB-PE and Activated_TB-PE showed 5750 DEGs, with 2669 genes upregulated and 3081 downregulated, further highlighting the modulatory role of TB-PE in shaping immune responses. Lastly, the comparison between the Activated and Activated_TB-PE conditions identified 282 differentially expressed genes (103 upregulated and 179 downregulated) Figure 2, suggesting that TB-PE may continue to modulate gene expression patterns following T cell activation, though the extent of change appears reduced. These findings underscore the critical role of TB-PE in modulating the immune landscape, leading to both suppression and selective activation of vital immune pathways. The presence of 31 common genes across the five comparison groups, as indicated by the Venn diagram (Figure 2 and Table S6), highlights shared regulatory mechanisms that are likely central to the interplay between immune suppression and activation during HIV-1/TB co-infection
Key gene regulation in HIV-1/TB co-infection
The detailed examination of crucial DEGs provides further insights into the molecular mechanisms regulating immune responses in HIV-1/TB co-infection. Several genes, including FGR, SNHG12, FASLG, CXCR4, and PSMD1, demonstrated significant expression changes across the conditions, shedding light on their role in modulating immune activity (Table S6). FGR, a proto-oncogene belonging to the Src family tyrosine kinases, showed negative expression in the Control vs TB-PE comparison (log2 fold change: -1.95). This means the TB-PE microenvironment can suppress FGR expression, leading to more immune suppression in the affected persons (Figure 3). However, in the TB-PE vs. Activated comparison, FGR was upregulated (log2 fold change: 1.818), indicating that T cell activation reverses this suppression, possibly restoring immune function and promoting viral reactivation. The dynamic effect of FGR seems to be imperative for the regulation of the immune system during co-infection.
Similarly, SNHG12 (small nucleolar RNA host gene 12) was involved in RNA metabolism and regulation and downregulated in Control vs TB-PE (Log2 fold change: -1.241). Following activation of T-cells (TB PE vs activated), expression levels of SNHG12 were upregulated (log2 fold change 3.79), thus supporting the premise that T-cell activation restores its function, altering expression patterns of genes and assisting the immune response. This regulation pattern indicates that SNHG12 is sensitive to changes in the immune environment, making it a critical player in regulating responses during co-infection.
FASLG (Fas ligand), which is involved in inducing apoptosis, was upregulated in Control vs TB-PE (log2 fold change: 1.209), reflecting the activation of apoptotic pathways under TB-PE conditions. However, FASLG was markedly downregulated in TB-PE vs. Activated (log2 fold change: -2.374), implying that T cell activation suppresses apoptosis, favoring cell survival. This shift from promoting cell death to enhancing survival could play a significant role in viral latency and reactivation, where immune suppression is lifted, allowing for T cell survival and viral persistence (Figure 3). CXCR4 (C-X-C motif chemokine receptor 4), which plays a role in immune cell trafficking and serves as a co-receptor for HIV entry, was significantly downregulated in the Control vs. TB-PE comparison (log₂ fold change: -1.382). This downregulation may reflect altered chemotactic signaling and potential changes in cellular susceptibility to HIV under TB-PE conditions. In contrast, CXCR4 was upregulated in the TB-PE vs. Activated comparison (log₂ fold change: 2.545), suggesting that T cell activation may restore its expression. This upregulation could be associated with increased chemotactic potential and possibly greater susceptibility to HIV-1 entry, although functional outcomes require further validation. This dual role of CXCR4 in both immune regulation and viral infection highlights its significance in the context of HIV-1/TB co-infection.28
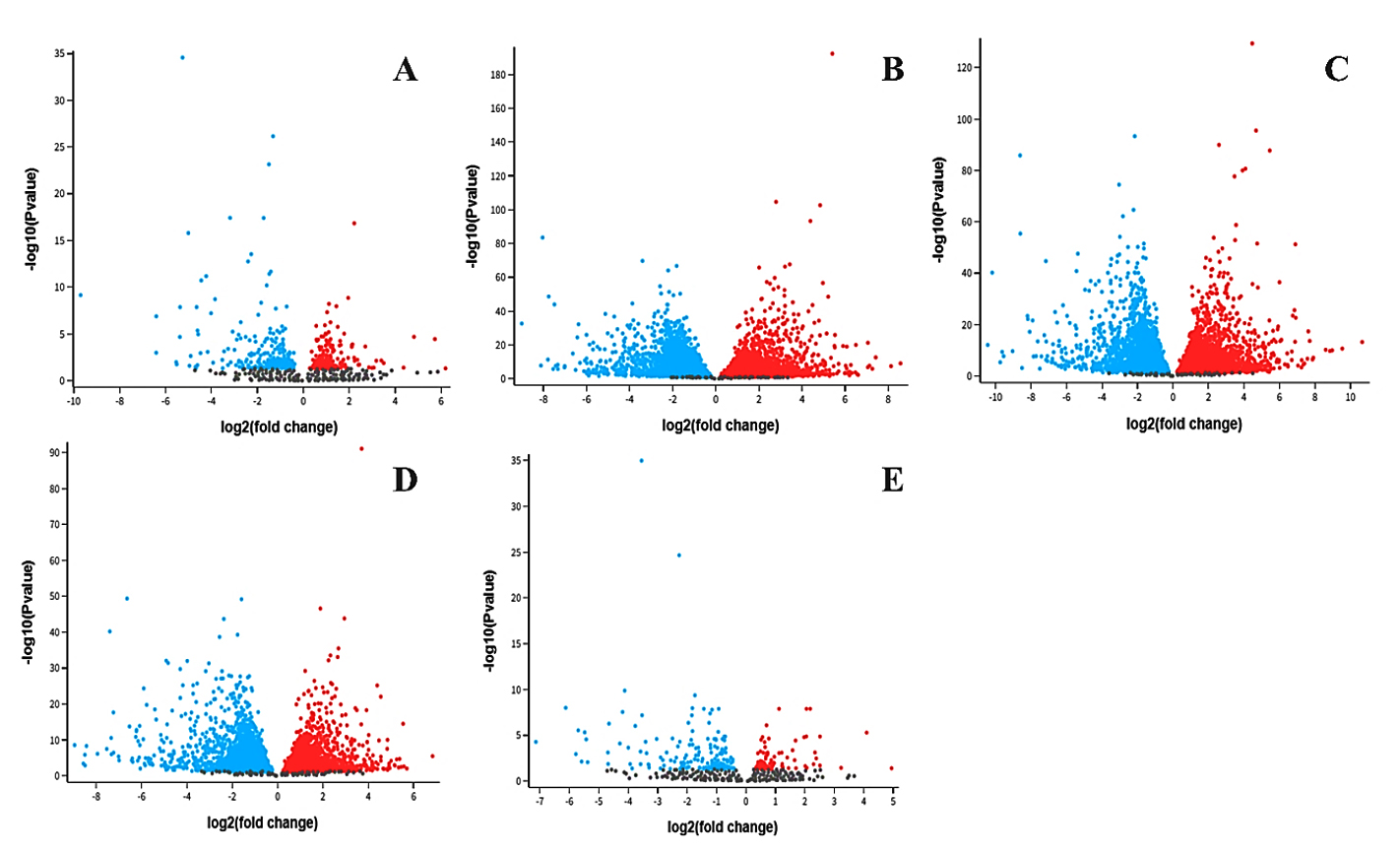
Figure 3. Volcano plots displaying differentially expressed genes across five comparison groups in the HIV-1/TB co-infection datasets: (A) Control vs. TB-PE, (B) Control vs Activated, (C) TB-PE vs. Activated, (D) TB-PE vs. Activated_TB-PE, and (E) Activated vs. Activated_TB-PE
PSMD1 (proteasome 26S subunit, non-ATPase 1) showed a more complex expression pattern, with a slight upregulation in TB-PE vs Activated (log2 fold change: 0.336) and significant downregulation in TB-PE vs Activated_TB-PE (log2 fold change: -1.818). PSMD1 is a protein degradation-related mechanism that regulates the immune response through viral and host protein turnover. This differential expression suggests that PSMD1 is vital for the homeostasis of cells during both immune suppression and reactivation by facilitating protein degradation with the changing immune environment. The configuration of these shared genes in different conditions reflects the dynamic control over immunological pathways in the case of HIV-1/TB co-morbidity. The changes in expression, especially regarding TB-PE and T-cell stimulation, provide critical molecular pathways that may be harnessed to develop therapies to alter immune responses to reduce viral reservoirs.
Cross-referencing common genes between HIV-1/TB and SARS-CoV-2
After identifying 31 common genes from the HIV-1/TB co-infection dataset using Venn diagram analysis of five distinct groups (Control, TB-PE, Activated, Activated_TB-PE), these genes were cross-referenced with genes associated with SARS-CoV-2 from the Comparative Toxicogenomics Database (CTD). The CTD database yielded 16,850 genes associated with COVID-19. Upon comparison, 27 common genes were identified between HIV-1/TB and SARS-CoV-2 infections (Figure 2 and Table S7). These shared genes highlight potential overlapping molecular pathways involved in immune regulation, viral persistence, and the host’s response to co-infection and viral infection. The 27 common genes identified between SARS-CoV-2 and HIV-1/TB include crucial regulators such as FASLG, SPON1, CXCR4, PSMD1, TAP1, and PIK3IP1, all of which play significant roles in immune signaling, apoptosis, and viral trafficking. These genes could represent key molecular players mediating the pathogenesis of SARS-CoV-2 and HIV-1/TB co-infection, offering insights into shared mechanisms of immune suppression and viral reactivation. Cross-referencing genes from two infectious diseases points to potential targets that could be explored for therapeutic intervention. This observation, as highlighted in the present analysis, allows addressing further questions about the potential roles of viral co-infections in inducing immune dysregulation and impacting disease progression.
Gene ontology and pathway enrichment analysis of genes between SARS-CoV-2 and HIV-1/TB co-infection
To understand the functional significance of the 27 common genes identified between SARS-CoV-2 and HIV-1/TB co-infection, we used the David module for Gene Ontology (GO) analysis and Pathway Enrichment analysis. According to GO analysis, the positive regulation of protein phosphorylation (GO:0001934, p-value = 0.000123) was indicated with five genes (DAB2, RALB, GPNMB, PELI2, AXIN2) as a crucial biological process (Figure 4). This process is central to immune signaling and modulation of cellular interactions, which is significant in the immune response to viral infections, such as SARS-CoV-2 and HIV-1/TB co-infection. Another essential biological process was the positive regulation of cell migration (GO:0030335, p-value = 0.0055), in which FGR, DAB2, GPNMB, and CXCR4 are involved (Table S8). Furthermore, cell migration is an essential need for the recruitment of immune cells to the infection sites for both pathogen clearance and the integration of immune responses against the co-infection.
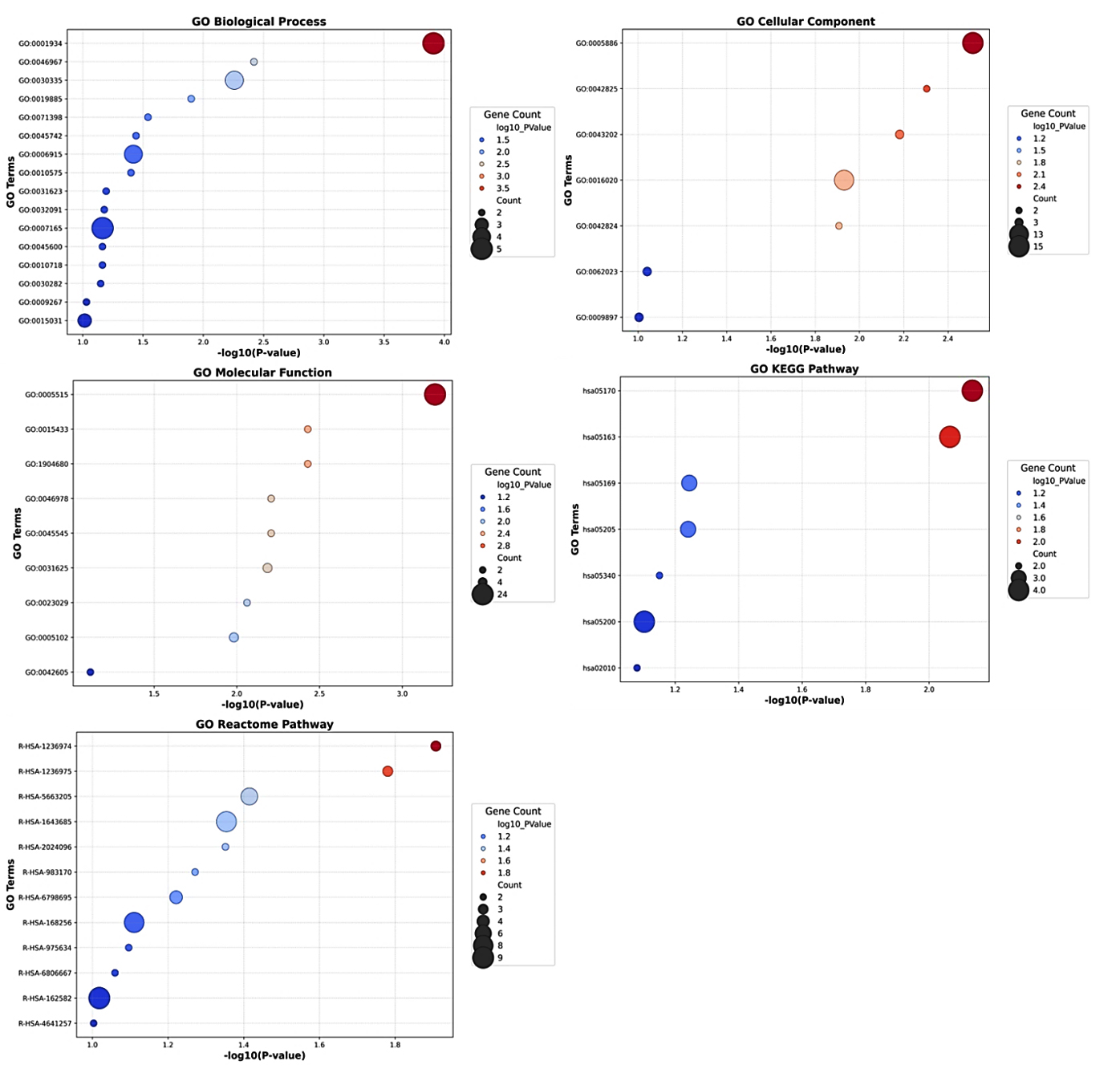
Figure 4. Gene Ontology and Pathway Enrichment Analysis of genes between SARS-CoV-2 and HIV-1/TB co-infection. The size of the circles indicates the number of genes involved in each pathway, with larger bars representing greater gene involvement in specific pathways
Additionally, the analysis revealed enrichment for antigen processing and presentation via MHC class I (GO:0019885, p-value = 0.0125), driven by the TAP1 and TAP2 genes. It is essential in activating cellular adaptive immune responses, especially the activation of cytotoxic T-cells to eliminate viral pathogens during co-infections. The cellular component enrichment analysis revealed several significant associations with the plasma membrane (GO:0005886, p-value = 0.0030) comprising 15 genes, including RALB, SDC2, C5AR1, TAP1, CXCR4, etc. This suggests that many identified genes are involved in membrane-associated processes integral to cell signaling and interaction with external pathogens. Also, the TAP complex (GO:0042825, p-value = 0.0049), which enriches TAP1 and TAP2, suggests why these genes are essential in processes involved in antigen presentation. This is very important in identifying viral antigens tissue and cells that contain viral persistence and immune evasion strategies employed by HIV and Mtb.
Moreover, the lysosomal lumen (GO:0043202, p-value = 0.0065), represented by genes such as SDC2, FASLG, and HPSE, stresses the necessity of lysosomal degradation in antigen processing. Lysosomes play a crucial role in breaking down pathogens and presenting their antigens to the immune system, thus initiating an effective immune response. The GO molecular function analysis revealed that the term with the highest enrichment was protein binding (GO:0005515, p-value = 0.0006), executed by 24 out of 27 genes (88.88%). This function is important for regulating viruses’ immune response, signaling, and replication (Figure 4). Genes such as LRRC25, SPON1, RALB, and FASLG are crucial in protein-protein interaction/mediated processes that are required to effectively coordinate the immune response towards a viral infection. In addition, it has been shown that ABC-type peptide antigen transporter activity (GO:0015433, p-value = 0.0037) and peptide transmembrane transporter activity (GO:1904680, p-value = 0.0037) exemplified by TAP1 and TAP2 are involved in the transport of molecules for the presentation of antigens on the cell surface. These activities are essential as they are vital in ensuring that any viral antigens taken up are accurately processed and displayed on the cell surface for the immune system to identify and attack the infected cells.
The KEGG pathway analysis was useful in revealing the role of some notable pathways, such as Human immunodeficiency virus one infection (hsa05170, p-value = 0.0073) and Primary immunodeficiency hsa05340 (p-value = 0.0706). These pathways included the genes TAP1, TAP2, CXCR4, and FASLG, all suggesting a possible role in viral immune evasion and persistence, especially in co-infection scenarios. In the Reactome pathway analysis, ER-Phagosome pathway (R-HSA-1236974, p-value = 0.0123) and Antigen Processing-Cross presentation (R-HSA-1236975, p-value = 0.0165) were significant, involving TAP1, TAP2, and PSMD1. These pathways are vital in presenting viral peptides for effectively killing T-cells against HIV-1 and Mtb. Related pathways such as Infectious disease (R-HSA-5663205, p = 0.0384) and Immune System (R-HSA-168256, p = 0.0775) were also found to be enriched, which further suggests that these common genes have a broader role in the control of responses to viral and bacterial challenges (Figure 4 and Table S8). The genes FGR, SDC2, CD9, and PSMD1 could contribute to immunity by playing significant roles in controlling both viral and host defenses, potentially affecting viral replication.
Functional annotation clustering and enrichment analysis
To investigate the biological roles found in the common genes between HIV-1/TB and SARS-CoV-2 infections, we performed functional annotation clustering for biological processes, molecular functions, or cellular component enrichment (Table S9). This analysis clarified the possible similarities in molecular pathways that are common in managing infections. These circumstances may be helpful for a better understanding of viral persistence, evasion of the immune response, and co-infection. Looking into the enriched terms and the gene clusters associated with them, we were interested in the biological function and cellular distribution of these genes concerning changing immune response and the virus replication cycle (Figure 5). The first cluster, with an enrichment score of 1.72, highlights terms such as membrane (GO:0016020), transmembrane helix, and extracellular region. Notable genes like LRRC25, SDC2, TAP2, TAP1, CXCR4, FASLG, and PSMD1 are highly involved in membrane-associated processes (Table S9). Panel A shows that many of these genes are positively associated with membrane and extracellular domain functions (green areas), underscoring their roles in immune responses and viral-host interactions. However, the black areas reveal gaps where gene associations for terms such as “Transmembrane Helix” and “Topological Domain: Extracellular” remain underexplored. The second cluster, with an enrichment score of 1.30, focuses on signaling receptor binding (GO:0005102) and the extracellular space. Genes such as FGR, PIK3IP1, LPL, and FASLG are significantly enriched in receptor-binding functions, crucial for immune modulation and signal transduction during viral infection. As illustrated in Panel B, these genes are predominantly associated with receptor binding and extracellular functions, reinforcing their importance in immune signaling. However, the black gaps indicate that certain associations, especially those involving the extracellular region, are underexplored.
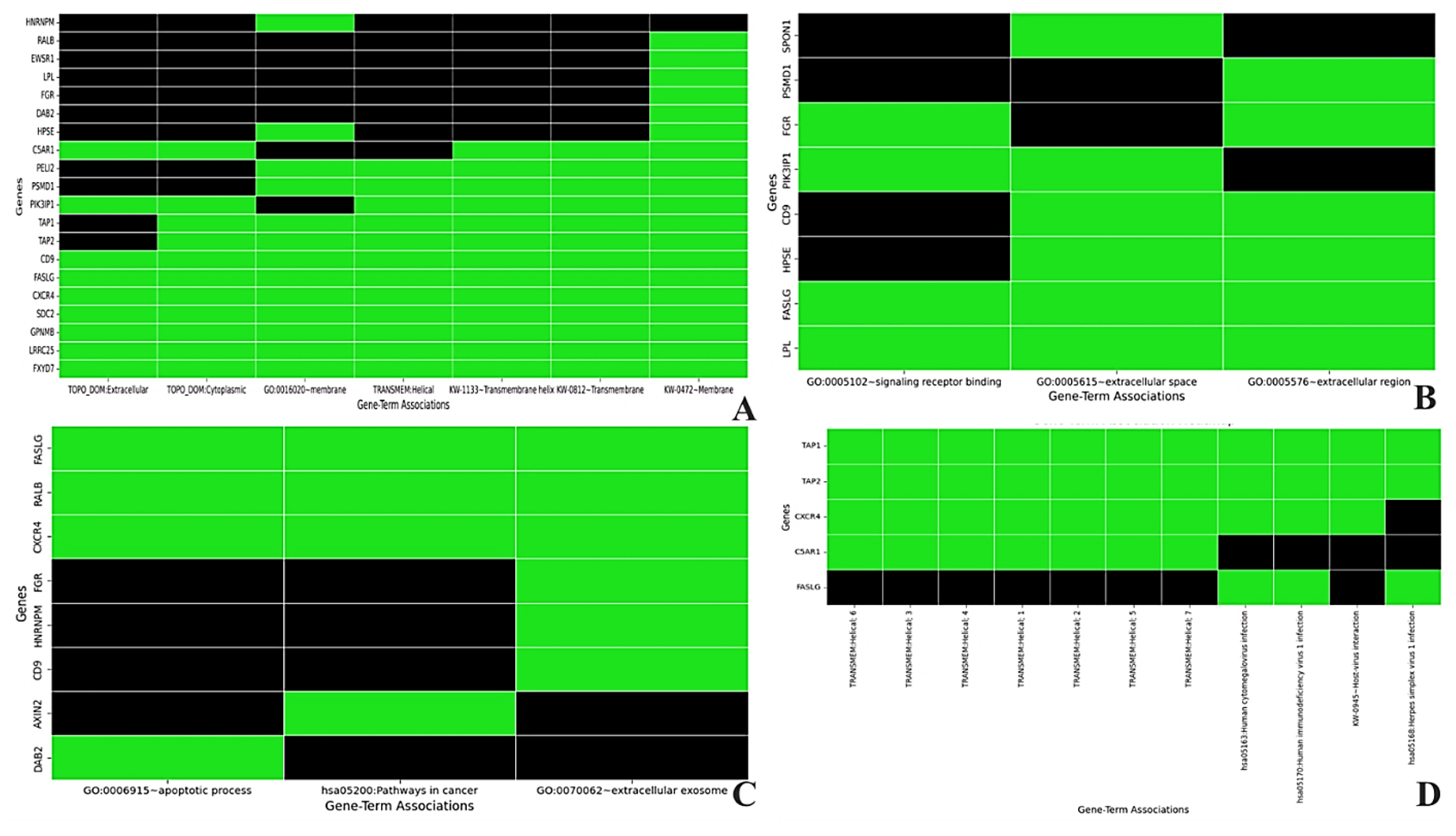
Figure 5. Functional-annotation clustering of shared genes into four clusters. Panel A shows cluster 1, Panel B cluster 2, Panel C cluster 3, and Panel D cluster 4. In all panels, green marks gene-term associations that have been positively reported in the literature, and black denotes associations not yet reported
The third cluster, with an enrichment score of 1.12, includes genes involved in the apoptotic process (GO:0006915), such as DAB2, CXCR4, RALB, and FASLG. These genes play essential roles in programmed cell death, critical for regulating immune responses and controlling viral infections by eliminating infected cells. As shown in Panel C, these gene-term associations are positively reported, underscoring the role of apoptosis in managing co-infections. However, unreported associations suggest further areas for investigation. The fourth cluster, with an enrichment score of 1.07, focuses on pathways related to Human Immunodeficiency Virus 1 infection (hsa05170) and cytomegalovirus infection (hsa05163), involving genes such as TAP2, TAP1, CXCR4, and FASLG. These genes are pivotal in viral infection pathways, particularly in modulating immune responses and processing viral antigens. Panel D highlights their strong association with viral pathways, particularly in antigen processing and immune evasion by viruses like HIV-1 and SARS-CoV-2 (Figure 5). Thus, this functional annotation clustering provides valuable insights into how shared molecular pathways contribute to immune regulation, viral persistence, and host response in co-infected individuals.
Insight from gene set enrichment in HIV-1/TB and SARS-CoV-2 co-infection
To explore shared molecular mechanisms and pathways between HIV-1/TB co-infection and SARS-CoV-2 infection, we analyzed multiple gene set libraries using term frequency-inverse document frequency (TF-IDF) values followed by UMAP for dimensional reduction. This analysis identified clusters of enriched gene sets, highlighting common regulatory pathways (Table S10). Figure 6 provides a visualization of the gene sets, with clusters color-coded based on their significance, illustrating transcription factor-related gene sets, pathways, disease associations, and cell types. The clustering of these gene sets reveals significant molecular interactions that could contribute to immune evasion, viral persistence, and co-infection dynamics. The analysis began by identifying transcription factor-related gene sets, shown in panels A-C of Figure 6. Cluster 2, highly significant in the ChEA 2022 dataset (panel A), included transcriptional regulators such as SUZ12, RNF2, and ATF6B, linked to genes like SPON1, MAFB, CXCR4, and HPSE. These transcription factors are essential in modulating gene expression during viral infections and immune responses. SUZ12 and RNF2, involved in chromatin remodeling, impact the host’s response to viral infections. Their enrichment in both HIV-1/TB and SARS-CoV-2 infections highlights their critical role in viral persistence and immune suppression, making them potential therapeutic targets.
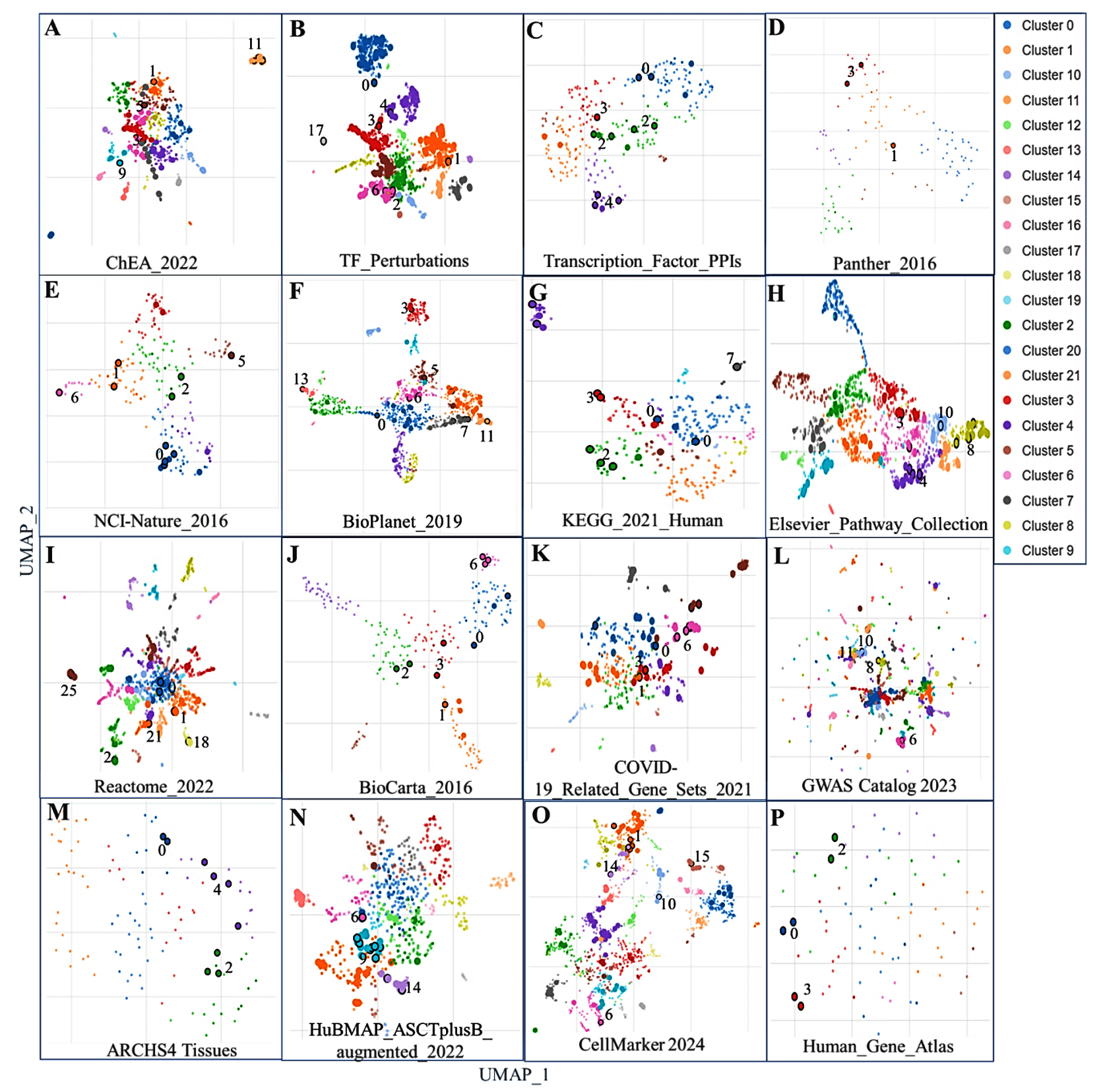
Figure 6. Scatterplot representing terms from a specific gene set library. Term frequency-inverse document frequency (TF-IDF) values were calculated for each gene set, followed by UMAP for dimensional reduction. The terms are displayed based on the first two UMAP dimensions, with similar gene sets appearing closer together. Colors represent clusters identified using the Leiden algorithm, applied to the TF-IDF values. Larger and darker points indicate terms with higher enrichment significance. Hovering over a point reveals the term, its enrichment p-value, and its assigned cluster. Panels A-C highlight transcription factor-related DEGs, D-J show various annotated pathways, K-L represents disease-associated genes, and M-P highlights cell types and gene sets from cell markers
Next, we explored pathway enrichment, focusing on common antigen presentation pathways, immune response, and viral infections. Gene cluster 7 of the KEGG 2021 Human database (panel G) depicts the contribution of TAP1, TAP2, CXCR4, and FASLG, essential in antigen processing and presentation, a hallmark in any immune response. The disruption of this process has crucial implications in the phenomenon of immune evasion which is commonly observed in HIV-1 and SARS-CoV-2 infections.29 Enriching antigen presentation genes in both infections suggests that improving this immune function could help develop therapies that enhance immune recognition and virus elimination. Particular pathways, such as human immunodeficiency virus one infection and ABC transporters, were significantly enriched, and this suggests potential molecular pathways that these viruses may utilize to manipulate the host immune response. Immune evasion mechanisms were particularly evident in Cluster 5 of the Elsevier Pathway Collection (Panel H) showed strong enrichment for genes such as TAP1, TAP2, and FASLG, which are associated with antigen presentation and immune evasion mechanisms. These genes have also been implicated in CXCR4-associated signaling cascades involved in HIV-1 entry and may play a similar role in SARS-CoV-2 infection, although this potential functional overlap requires further investigation. The finding poses a possibility of co-infections taking advantage of these immune pathways to make the immune system more suppressed; hence, the disease becomes complicated. The two viruses implemented the same immune evasion, suggesting plausible considerations in administering these viruses. In addition to pathways, disease-associated gene sets were explored in panels K and L, where Cluster 9 was highly significant. This cluster consisted of immunologically essential genes FGR, MAFB, and CXCR4 in HIV-1/TB and SARS-CoV-2 infection. For example, it is well known that CXCR4, which is expressed on activated T cells and is important in recirculating T cells, is also involved in immunological aberrations in both infections. These examples also indicate that unique gene sets amplify the comorbid conditions of disease and, therefore, share therapeutic endpoints.
In addition, further examination of the gene sets in specific cell types gave further information about the immunological landscapes of these infections. Panels M-P showed strong enrichment of macrophage, dendritic cell, and monocyte-associated genes, which are vital contributors in the first line of innate defense. Cluster 3, especially abundant in the ARCHS4 Tissues data set (panel M), consisted of genes such as LRRC25, CXCR4, and HPSE, all of which undergo high expression in macrophages and monocytes. These cell types play an early role during infection by responding to pathogens and eliciting an immune response against the pathogen. Enriching immune cell-specific gene sets suggests their crucial role in orchestrating the immune response to HIV-1/TB and SARS-CoV-2, mainly through cytokine production and antigen presentation. This underscores the importance of these cell types in managing viral infections and their potential as targets for therapies aimed at boosting innate immunity. Across the various gene set libraries, several clusters consistently pointed to immune modulation, viral entry, and immune escape mechanisms shared between HIV-1/TB and SARS-CoV-2. Cluster 1, identified in several datasets (panels F, I, and N), showed significant involvement of antigen processing and immune signaling pathways, highlighting the interplay between viral infections and immune suppression. The co-enrichment of genes like TAP1, TAP2, and CXCR4 suggests that these genes are central to the immune evasion strategies used by both viruses. Identifying these overlapping pathways opens up new avenues for developing broad-spectrum therapies that target immune regulation in viral co-infections.
Insights from Protein-Protein Network Interaction Analysis
After identifying the shared genes between HIV-1/TB and SARS-CoV-2 infections, we performed a protein-protein interaction (PPI) network analysis to explore the functional relationships among these genes, aiming to uncover critical molecular pathways and regulatory hubs involved in these infections (Figures 7 and 8). The final merged network, as shown in Figure 7, consists of 57 nodes and 153 edges, illustrating the complex molecular interactions that drive the progression of both infections. The clustering coefficient 0.118 reflects moderate clustering of functional gene groups, while the characteristic path length of 1.032 suggests that the network is highly efficient, with most nodes being connected through short paths (Table S11). The network’s diameter of 2 and radius of 1 further emphasize the compact and highly interconnected structure, with hub genes such as TAP1, TAP2, and CXCR4 playing central roles in immune regulation and viral evasion (Figure 7).
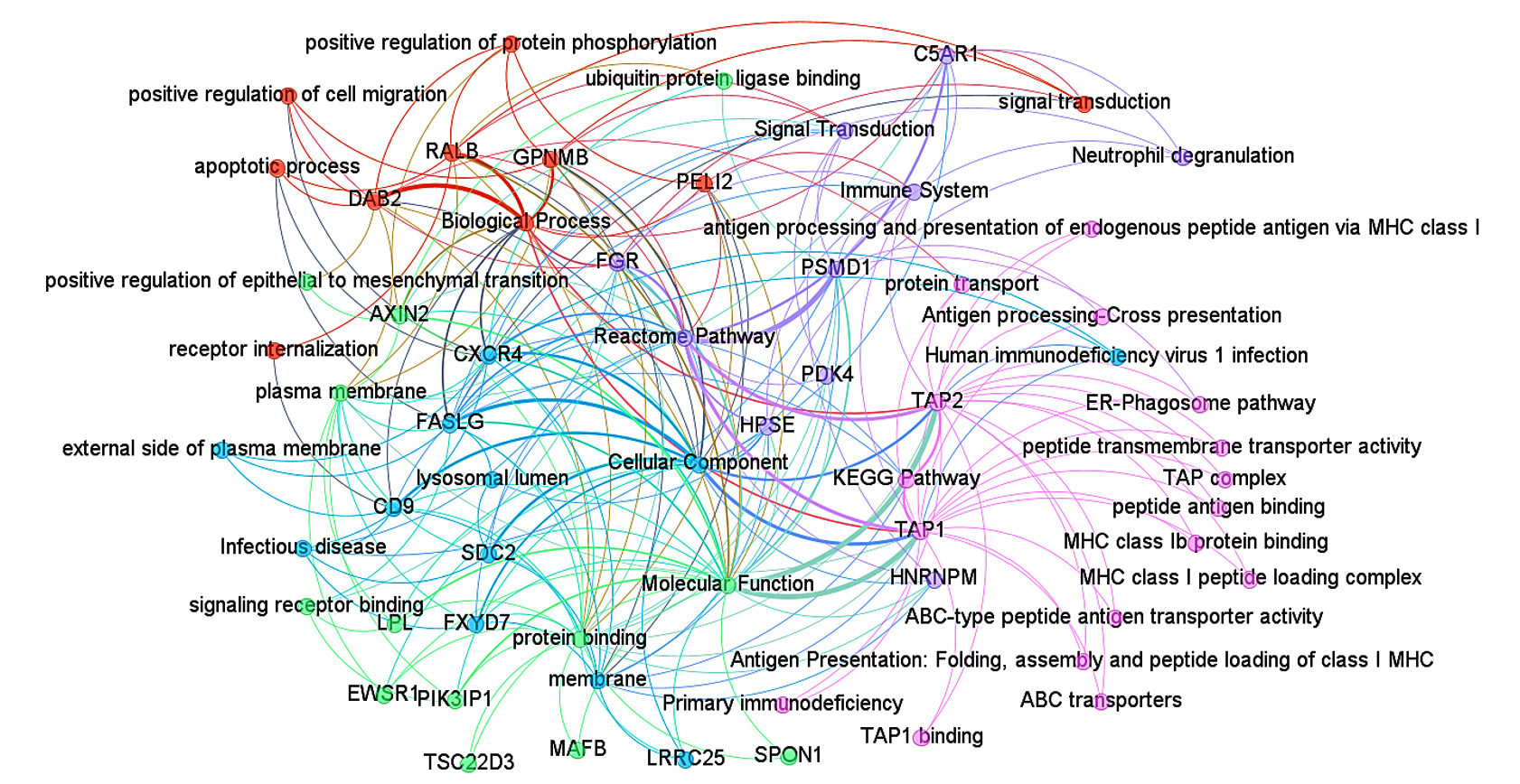
Figure 7. Protein-protein interaction network analysis of the overall pathways. The nodes and edges are color-coded to represent different functional annotations: green for molecular function, blue for cellular components, pink for KEGG pathways, orange for biological processes, and violet for Reactome pathway analysis
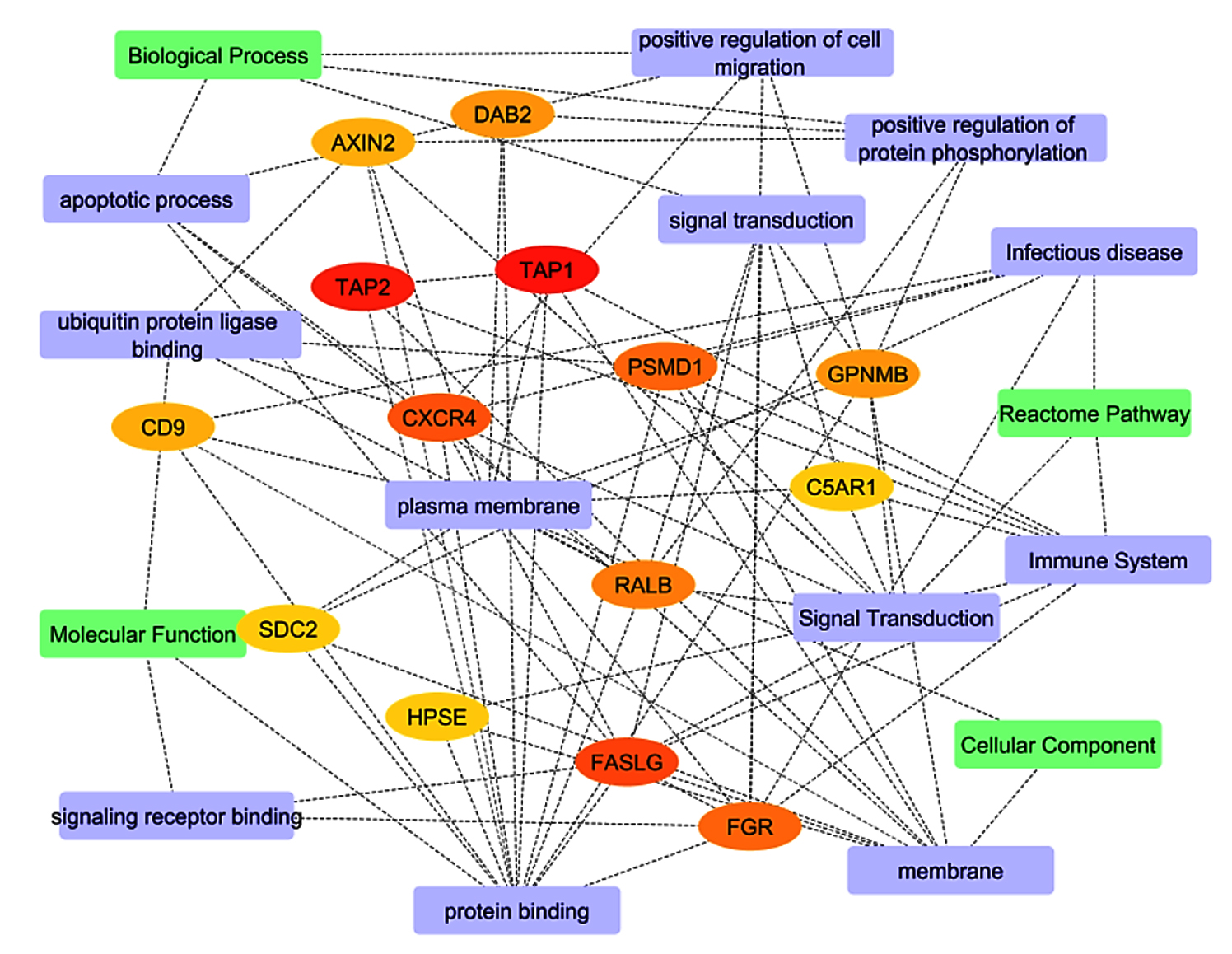
Figure 8. Top hits in the protein-protein interaction network within pathways. Green represents pathways based on degree selection, violet indicates specific pathways and other colors highlight the genes associated with those pathways
Central hub genes, including TAP1, TAP2, and CXCR4, were identified based on their degree of interaction within the network (Table S12). These genes play an important role in antigen processing and presenting (TAP1, TAP2) and in immune-cell trafficking (CXCR4), which is critical in both activation of the immune system and entry of viruses. Plasma membrane processes, embedded with a rank of 16, indicate these functions towards receptor-triggered immune activation. FASLG, another hub gene, is vital in inducing apoptosis in infected cells (score 12), which is essential in bringing down viral multiplication. Likewise, PSMD1 and FGR (with scores of 9) participate in immune signaling and regulation, mainly in antigen processing and signal transduction, which supports the idea of their functional importance in immune response pathways (Figure 8). The network structure was then further explored by modularity analysis, which revealed core functional units of the network that are engaged in one or more biological processes, cellular, molecular functioning, and pathways. The outlined modularity classes are marked in different colors in Figure 7; class 2 (Pink, 27.42%) is a Modularity Class focused on antigen presentation and dependent on specific pathways in KEGG. It demonstrates how immune evasion strategies contributed by TAP1, TAP2, and CXCR4 lead to the persistence of infections like HIV-1 and SARS-CoV-2. Modularity 4 appeared in grains, targeting molecular functions such as protein binding and transporter activity. It was centered around genes PSMD1 and SDC2, crucial to cellular signaling and protein degradation and necessary for proper immune regulation. Modularity Class 3 (Sky Blue, 19.35%) emphasizes cellular components, particularly membrane-associated processes. CXCR4, HPSE, and FASLG are integral to receptor-mediated signaling and immune cell activation, critical for viral entry and cell trafficking.
Modularity Class 1 (Orange, 16.13%) focuses on biological processes such as apoptosis, signal transduction, and the positive regulation of protein phosphorylation. Genes like DAB2 and GPNMB play vital roles in immune cell survival, processes that viruses often manipulate to evade immune responses. Finally, Modularity Class 5 (Violet, 16.13%) encompasses Reactome pathways, with genes like C5AR1 and FASLG regulating inflammatory and immune signaling pathways. This cluster highlights the role of immune dysregulation in exacerbating co-infection dynamics. In the filtered protein-protein interaction network (Figure 8), we focused on the most significant interactions, further identifying TAP1, TAP2, and CXCR4 as central hub genes, with additional critical players like FASLG and PSMD1. These genes regulate crucial immune functions such as antigen presentation, viral entry, and immune escape, reinforcing their potential as therapeutic targets to enhance immune responses and facilitate viral clearance. The network’s structural properties reveal high connectivity, with an average of 5.368 neighbors per node. This suggests that hub genes like TAP1 and TAP2 interact with multiple other proteins and pathways, further emphasizing their regulatory importance. The compact nature of the network, indicated by short path lengths, suggests that functional signals can spread efficiently between genes, a hallmark of networks involved in immune responses and viral infections.
Tissue-specific and single-cell gene expression analysis in co-infections
To identify essential tissues involved in immune regulation and mechanisms of viral persistence during HIV-1/TB and SARS-CoV-2 co-infections, we analyzed gene expression profiling. Expression patterns of critical functional genes in the liver, lungs, spleen, and whole blood were depicted in the heat map (Figure 9) and are responsible for both local and systemic immune responses. In these tissues, a ‘core’ pattern that includes elevated expression of CXCR4, TAP1, TSC22D3, and FGR was identified, implying these gene’s roles in immune evasion, T cell antigen presentation, and viral replication control. High TAP1, TAP2, PSMD1, and CXCR4 gene expression levels were observed in whole blood and spleen, suggesting that these genes are pivotal in immunological signaling and MHC-mediated antigen presentation. This means that these tissues are crucial for body immunological responses to co-infections, as timely processing of viral antigens is essential for eliminating the pathogens. The upregulation of TAP1 and TAP2, essential components of the MHC class I antigen presentation pathway, may represent the immune system’s response to counteract the immune evasion strategies employed by HIV-1 and SARS-CoV-2.
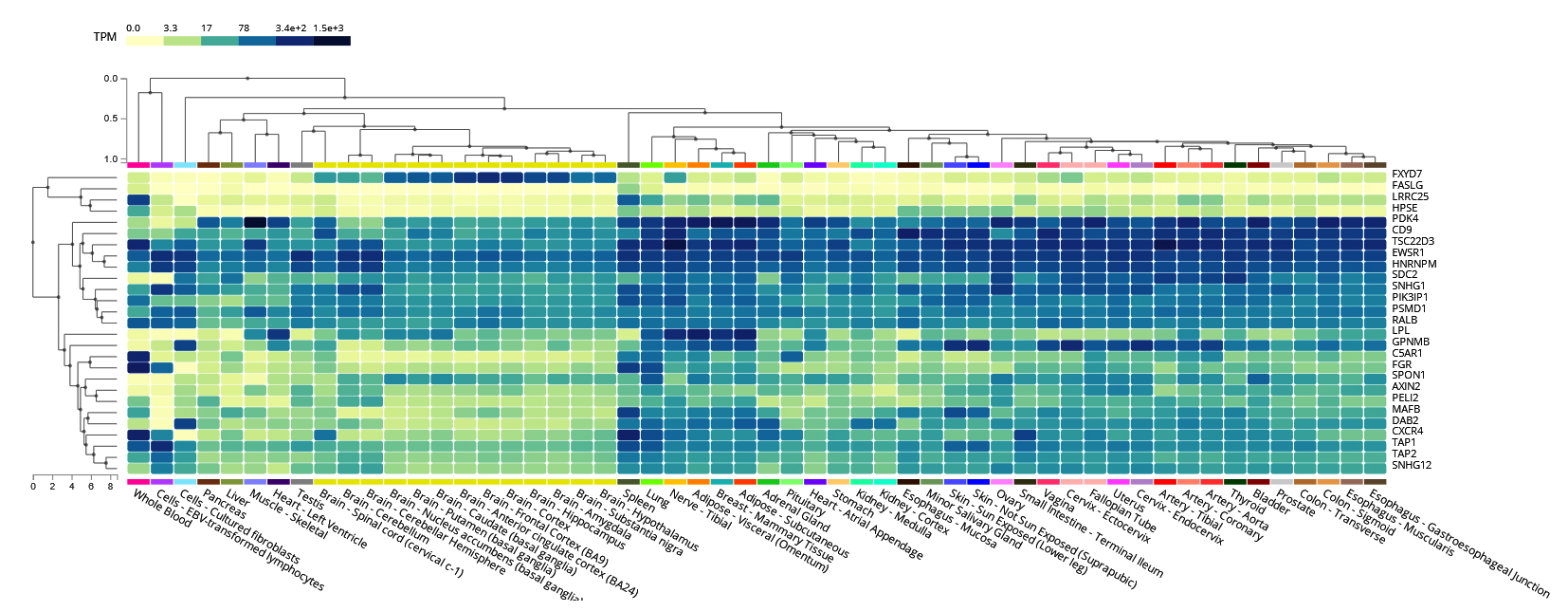
Figure 9. Cluster analysis of tissue-specific expression patterns of a common gene across co-infections. The analysis highlights variations in gene expression levels in different tissues during co-infection scenarios
Furthermore, CXCR4, a known co-receptor for HIV-1 and possibly of critical importance in SARS-CoV-2 pathogenesis, was highly expressed, suggesting its involvement in entering viruses into immune cells, leading to chronic infection. In the lung tissue, however, the expression of CXCR4 and FGR was also profoundly high, implying that the lungs are the main host for the movement of the immune cells and the entry of the virus. FGR, which is expressed in immune cells such as macrophages, may be involved in immune activation and viral clearance, suggesting the lung as a critical site for immune response modulation in these co-infections. Similarly, the detection of HNRNPM in lung tissue points to a potential role in post-transcriptional regulation, with possible implications for viral replication and immune response modulation. While the liver displayed relatively lower expression levels for most immune-related genes, SPON1 and AXIN2 were notably expressed, suggesting that the liver, although not a primary immune organ, plays a role in inflammation and tissue remodeling during viral co-infections (Figure 9). These genes manage metabolic disturbances and inflammation, indicating the liver’s contribution to broader immune-metabolic regulation.
In addition to the tissue-level analysis, a multi-gene single-cell analysis (Figure 10) provided further insight into the cellular-level expression of critical genes within the lung, an essential organ in managing immune responses during viral infections. TSC22D3 exhibited significant expression across several immune cell types, particularly mast cells (49.12%) and T cells (31.97%), indicating its potential role in modulating immune responses, especially in regulating inflammation and immune suppression. This gene’s activity is critical for balancing immune responses in viral infections, reducing hyperactivation that could lead to immunopathology. CXCR4, highly expressed in T cells (32.14%), reinforces its known role as a receptor in immune cell trafficking and viral entry. Given that CXCR4 is a known co-receptor for HIV-1, it likely facilitates similar viral interactions in SARS-CoV-2 infections, influencing viral persistence in immune cells. FGR was notably expressed in alveolar macrophages (30.7%), highlighting these cells’ crucial role in lung pathogen recognition and immune cell activation. Alveolar macrophages are critical for viral clearance and immune defense in respiratory infections, which aligns with the gene’s expression profile during co-infections. This suggests that alveolar macrophages may be critical mediators in the localized immune response within the lung during co-infections of HIV-1/TB and SARS-CoV-2. Lastly, SPON1 showed significant expression in lung fibroblasts (46.54%), which are involved in tissue remodeling and repair (Figure 10). The upregulation of SPON1 in fibroblasts may indicate a role in fibrosis and tissue repair, particularly in severe co-infection scenarios involving extensive lung damage. This observation is consistent with existing knowledge of fibroblasts contributing to tissue remodeling and viral damage management in chronic and severe infections.
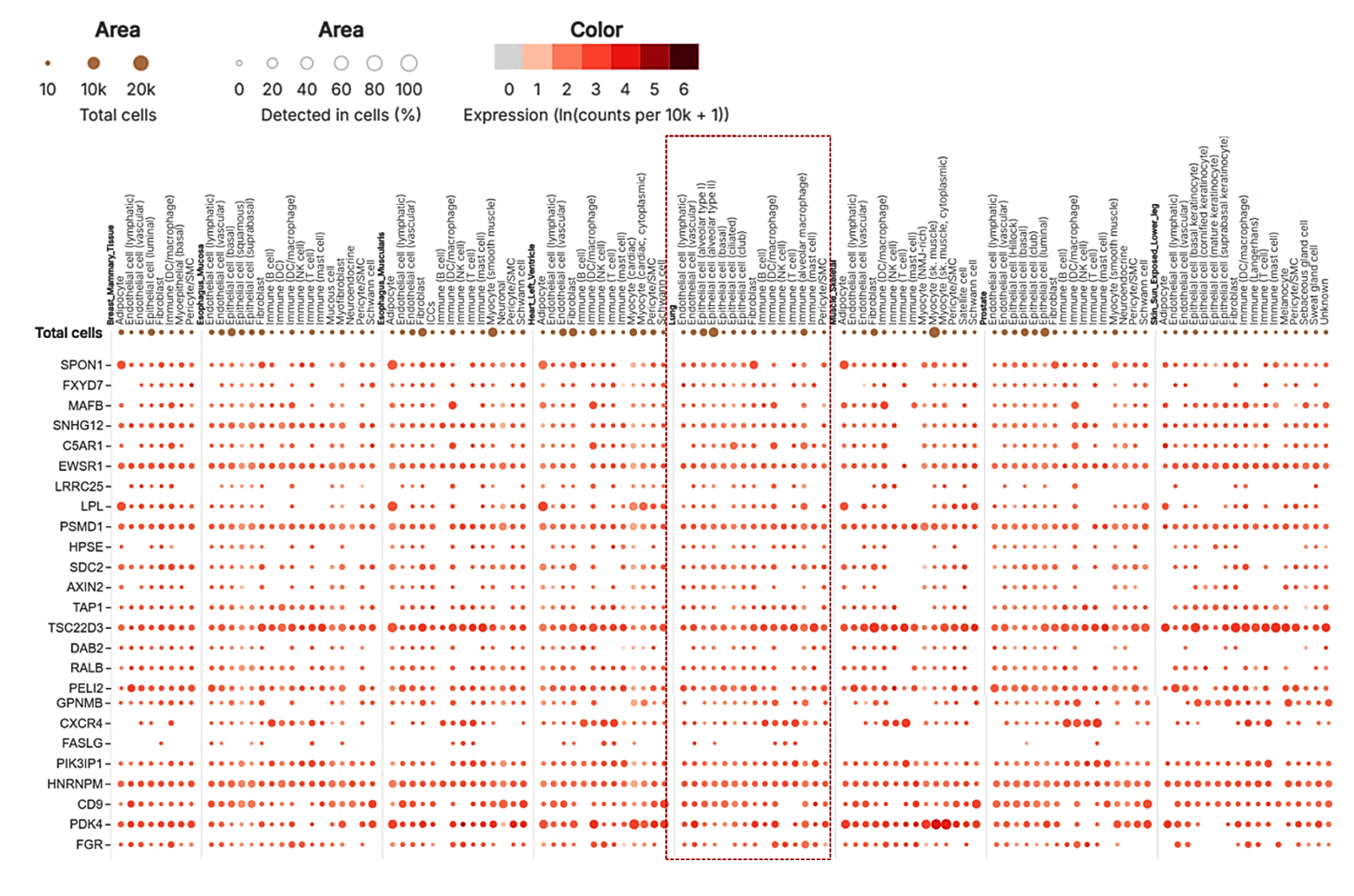
Figure 10. Single-cell RNA sequencing analysis, with the dotted box specifically highlighting lung tissue. The analysis provides detailed insights into gene expression at the single-cell level within the lung tissue
The interrelation between HIV-1 and Mtb complex, TB, causes one of the persistent global health concerns due to their effects on immunity, which is less understood, leading to increased viral persistence and ability to suppress immunity. Since the emergence of the novel coronavirus SARS-CoV-2, the immune situation in such co-infected individuals has become even more complicated because similar molecular pathways of these pathogens can contribute to a more severe disease. Understanding these shared mechanisms is critical for identifying therapeutic targets and improving treatment outcomes for patients with viral co-infections. Our study aimed to explore the molecular pathways and immune regulation mechanisms linked to these co-infections by performing an integration of gene expression profiling, and single-cell multi-gene analysis.
Gene expression studies performed in the presence of various conditions like Control, TB-PE, Activated, and activated_TB-PE demonstrated the critical mechanisms involved in HIV-1/TB co-infection. TB-PE led to considerable changes in the levels of immune-responsive genes, where some genes were upregulated while some were downregulated. Of the comparisons made, the most pronounced number of differentially expressed genes (DEGs) was recorded for TB-PE vs. Activated, indicating the active mechanism whereby T cell activation can reverse TB-PE-associated immunosuppression. Other genes such as FGR, SNHG12, FASLG, and CXCR4 were found to be dynamically regulated, further evidencing that they aid in regulating immune activity and controlling viral persistence. These changes in gene regulation reveal the detailed interaction between suppression and activation of the immune system during co-infection with HIV-1 and TB, where viral activity in a latent state and immune response mechanisms shape the immune environment continuously. We conducted further analysis by identifying 27 common genes between HIV-1/TB and SARS-CoV-2. We also performed several analyses of how the levels of expression of specific genes such as CXCR4, TAP1, TSC22D3, and FGR are correlated with specific tissues-the liver, lung, spleen, and blood-pivotal for local and systemic immunity. Some of the most expressed genes, TAP1, TAP2, PSMD1, and CXCR4, were also found in the spleen and whole blood and expressed in high levels, implying their antigen-presenting and signaling functions. Systemic immune responses are also crucial in handling co-infection with other pathogens, making it compulsory to process viral antigens for recognition and elimination. Increased CXCR4 and FGR expression in the lung tissue could further denote the organ’s role as a niche for the movement of immune cells and entry of viral particles. HNRNPM in lung tissues indicates a role in post-transcriptional processes connected with potential effects on viral persistency and immunity. The liver’s lower expression levels of immune-related genes were noted; however, proteins such as SPON1 and AXIN2 were quite active, evidencing inflammation or tissue remodeling by the liver during co-infections.
Further insight was obtained from the single-cell multi-gene analysis, which identified crucial immune modulation genes in different lung cells. TSC22D3 was abundantly expressed in mast and T cells, regulating immunological responses and inflammation. In T cells, CXCR4 expression is characteristic of such cell’s role in migration and entry of viruses to such cells, particularly with HIV-1 and SARS-CoV-2 co-infection. Alveolar macrophages were found to express FGR, which highlights the importance of these immune cells in activating immune responses and clearing pathogens in the lungs. The high expression of SPON1 in lung fibroblasts also indicates that it plays an active part in fibrosis and lung tissue remodeling during severe co-infections. Specific critical hubs such as TAP1, TAP2, and CXCR4 involved in antigen presentation and immune cell migration were identified by studying protein-protein interactions. These hubs were part of a highly connected large network with 57 nodes and 153 edges with evidence of modularity relative to clusters of function revealing immune evasion, protein binding, and signal transduction. The descriptions of these central hub genes point out their importance in maintaining immune balance and controlling counter-infection responses. Examining 31 common genes from the HIV-1/TB co-infection dataset and those from the SARS-CoV-2 dataset uncovered 27 overlapping genes, bringing to light common molecular mechanisms related to immune regulation, viral persistence, and immune evasion. Essential genes, including TAP1, CXCR4, FGR, and PSMD1, were enriched in pathways related to antigen presentation, immune signaling, and apoptosis. Functional and pathway enrichment analyses focused on protein phosphorylation, cell migration, and antigen processing and presented the data. The insights gained from these results relate to the association between viral persistence and host immune suppression in the context of HIV-1/TB and SARS-CoV-2 infections, identifying prospective therapeutic targets for treating these complicated infections.
In the context of co-morbidities, including HIV-1, TB, and SARS-CoV-2, there is a critical outlook that concerns common pathways of immune suppression and survival strategies employed by the viruses. Our study was conducted within a bioinformatic and systems biology framework involving gene expression profiling, protein-protein interaction networks, and multi-gene single-cell analysis. This facilitated the identification of critical regulatory genes, such as CXCR4, TAP1, TSC22D3, and FGR, that regulate processes like antigen presentation, immune system signaling, and viral entry. Results of systems biology approaches show how such pathogens induce immune imbalances and highlight the complex pattern of concurrent infections. A comprehensive integration of tissue-type and single-cell approaches underscored the implications of immune regulation at both systemic and cellular levels and revealed differential gene expression patterns in the lung, spleen, and whole blood. The comparison of strongly associated genes between HIV-1/TB and SARS-CoV-2 viruses focused on deeply interrelated genetic mechanisms of the pathogens, emphasizing the necessity for addressing both infections within combined therapeutic strategies. Further studies, predominantly in wet-lab and clinical domains, are necessary to confirm these results and probe the functional contributions of highlighted hub genes in regulating immune function. This work lays a groundwork for designing therapies that aim to determine the mode of action of immune responses and advance clinical results for people struggling with these demanding co-infections.
Additional file: Additional Table S1-S2 and Figure S1-S12.
ACKNOWLEDGMENTS
The authors extend their appreciation to Northern Border University, Saudi Arabia, for supporting this work through project number NBU-CRP-2025-2043.
CONFLICT OF INTEREST
The authors declare that there is no conflict of interest.
AUTHORS’ CONTRIBUTION
All authors listed have made a substantial, direct and intellectual contribution to the work, and approved it for publication.
FUNDING
This study was funded by Northern Border University, Saudi Arabia, vide project number NBU-CRP-2025-2043.
DATA AVAILABILITY
All datasets generated or analyzed during this study are included in the manuscript and in the GitHub repository: https://github.com/IMRANINDERLOK/tb-hiv-covid19-genomic-study.git
ETHICS STATEMENT
Not applicable.
- Imran M, Abida, Alotaibi NM, et al. QcrB inhibition as a potential approach for the treatment of tuberculosis: A review of recent developments, patents, and future directions. J Infect Public Health. 2023;16(6):928-937.
Crossref - Alsayed SSR, Gunosewoyo H. Tuberculosis: Pathogenesis, Current Treatment Regimens and New Drug Targets. Int J Mol Sci. 2023;24(6):5202.
Crossref - Manaithiya A, Bhowmik R, Bhattacharya K, et al. A cheminformatics and network pharmacology approach to elucidate the mechanism of action of Mycobacterium tuberculosis g-carbonic anhydrase inhibitors. Front Pharmacol. 2024;15:1457012.
Crossref - World Health Organization. Global Tuberculosis Report 2022. 2022. https://www.who.int/teams/global-tuberculosis-programme/tb-reports/global-tuberculosis-report-2022. Accessed February 19, 2025
- Lv T, Cao W, Li T. HIV-Related Immune Activation and Inflammation: Current Understanding and Strategies. J Immunol Res. 2021;2021(1):7316456.
Crossref - World Health Organization. HIV and Tuberculosis. https://www.who.int/westernpacific/health-topics/hiv-aids/hiv-and-tuberculosis. Accessed February 19, 2025
- Kwok AJ, Mentzer A, Knight JC. Host genetics and infectious disease: new tools, insights and translational opportunities. Nat Rev Genet. 2021;22(3):137-153.
Crossref - Lu YJ, Barreira-Silva P, Boyce S, Powers J, Cavallo K, Behar SM. CD4 T cell help prevents CD8 T cell exhaustion and promotes control of Mycobacterium tuberculosis infection. Cell Rep. 2021;36(11):109696.
Crossref - de Martino M, Lodi L, Galli L, Chiappini E. Immune Response to Mycobacterium tuberculosis: A Narrative Review. Front Pediatr. 2019;7:464617.
Crossref - Vijayan KKV, Karthigeyan KP, Tripathi SP, Hanna LE. Pathophysiology of CD4+ T-Cell depletion in HIV-1 and HIV-2 infections. Front Immunol. 2017;8:257162.
Crossref - Bell LCK, Noursadeghi M. Pathogenesis of HIV-1 and Mycobacterium tuberculosis co-infection. Nat Rev Microbiol. 2018;16(2):80-90.
Crossref - Cronin S, de Vries-Egan A, Vahlas Z, et al. The immunosuppressive tuberculosis-associated microenvironment inhibits viral replication and promotes HIV-1 latency in CD4+ T cells. iScience. 2024;27(7):110324.
Crossref - Moss P. The T cell immune response against SARS-CoV-2. Nat Immunol. 2022;23:186-193.
Crossref - Carabelli AM, Peacock TP, Thorne LG, et al. SARS-CoV-2 variant biology: immune escape, transmission and fitness. Nat Rev Microbiol. 2023;21(3):162-177.
Crossref - Hoft MA, Burgers WA, Riou C. The immune response to SARS-CoV-2 in people with HIV. Cell Mol Immunol. 2024;21(2):184-196.
Crossref - Nelson RW, Chen Y, Venezia OL, et al. SARS-CoV-2 epitope-specific CD4+ memory T cell responses across COVID-19 disease severity and antibody durability. Sci Immunol. 2022;7(73):eabl9464.
Crossref - Waters R, Ndengane M, Abrahams MR, Diedrich CR, Wilkinson RJ, Coussens AK. The Mtb-HIV Syndemic Interaction: Why Treating M. tuberculosis Infection May Be Crucial for HIV-1 Eradication. Future Virol. 2020;15(2):101-126.
Crossref - Zebley CC, Zehn D, Gottschalk S, Chi H. T cell dysfunction and therapeutic intervention in cancer. Nat Immunol. 2024;25:1344-1354.
Crossref - Moine-Franel A, Mareuil F, Nilges M, Ciambur CB, Sperandio O. A comprehensive dataset of protein-protein interactions and ligand binding pockets for advancing drug discovery. Sci Data. 2024;11(1):402.
Crossref - Edgar R, Domrachev M, Lash AE. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30(1):207-210.
Crossref - Davis AP, Wiegers TC, Johnson RJ, Sciaky D, Wiegers J, Mattingly CJ. Comparative Toxicogenomics Database (CTD): update 2023. Nucleic Acids Res. 2023;51(D1):D1257-D1262.
Crossref - Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28(1):27-30.
Crossref - Kuleshov MV, Jones MR, Rouillard AD, et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44(W1):W90-7.
Crossref - Szklarczyk D, Gable AL, Lyon D, et al. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47(D1):D607-D613.
Crossref - Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13(11):2498-2504.
Crossref - Heymann S. Gephi. In: Alhajj R, Rokne J (Eds) Encycl Soc Netw Anal Min. Springer. NY. 2017:1-14.
Crossref - The GTEx Consortium. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science 2020;369(6509):1318-1330.
Crossref - Yusuf Aliyu A, Adeleke OA. Latest Progress on Tuberculosis and HIV Co-Infection: A Closer Look at People of Different Ages. Adv Ther. 2024:2400033.
Crossref - Chen B, Julg B, Mohandas S, Bradfute SB, Recover Mechanistic Pathways Task Force. Viral persistence, reactivation, and mechanisms of long COVID. Elife. 2023;12:e86015.
Crossref
© The Author(s) 2025. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.