


ISSN: 0973-7510
E-ISSN: 2581-690X
http://dx.doi.org/10.22207/JPAM.13.1.19 | © The Author(s). 2019
MAS, which is currently grouped under secondary or acquired haemophagocytic lymphohistiocytosis (sHLH), is a rare and fatal disorder that results from excess activation of T-cells and macrophages. Though the pathogenesis of MAS is poorly understood, various proinflammatory cytokines like interleukins (IL-1, IL-6), tumor necrosis factor a (TNF a), interferons are thought to play significant roles. MAS is associated with various clinical features such as non-remitting fever, bleeding, cytopenias, splenomegaly, hepatic dysfunctions, increased levels of triglyceride, ferritin and decreased levels of albumin and fibrinogen. Early diagnosis and interventions are crucial to reduce mortality risk but diagnosis is not often easy due to persistence of wide range of features that overlap with other rheumatic diseases, most commonly sJIA (systemic juvenile idiopathic arthritis). Corticosteroids and cyclosporins are commonly used for MAS treatment. Intravenous immunoglobulins, biologic agents like IL-1 blockers (anakinra, canakinumab), IL-6 blockers (tocilizumab) are also frequently used. Moreover there is still the need of genetic and immunohistological study in order to understand the exact mechanism of the syndrome development and establishment of novel therapies with lesser toxicities.
MAS, haemophagocytic lymphohistiocytosis, systemic juvenile idiopathic arthritis, cytokines, corticosteroids.
Various autoimmune and inflammatory states are known to be associated with a number of life threatening conditions. One of them is Macrophage activation syndrome1, that is more commonly observed in rheumatic diseases such as systemic juvenile idiopathic arthritis (sJIA), Kawasaki disease, systemic lupus erythro-matosus (SLE), Still’s disease, juvenile dermato-myositis and Becket’s disease2,3,4.
MAs does not hold a recent history. Familial lymphohistocytosis that shares quite similar clinical picture to MAS was first observed in 19525. Later, Boone in 1976, reported MAS among the patients with compulsive coagulo-pathy and hepatic insufficiency5 but Stephen et al was the one who coined the term Macrophage activation syndrome in 1993 and demonstrated that the excess activation of macrophages in most of his patients6. MAS that develops as a result of excess activation of macrophages, is emerging as a well characterized clinical facet that may be associated with different drugs, infections, cancers, hematological and rheumatic disorders or any isolated disease occurring with or without trigger7.
Prevalence
Though a rare complication, MAS has become more common these days. In a study from tertiary care hospital about 6.7% cases of sJIA had MAS8. MAS is more frequently observed in children but increased prevalence is also present in adults nowadays9. It is a life threatening disorder with mortality rate of 20-30%10. Among the rheumatologic disorders, sJIA is most commonly associated with MAS11 with the prevalence rate of 7-13%12, but the rate may vary upto 30-40% in the unreflected cases of MAS13. According to Gormezano et al, MAS is also frequently associated with SLE in children with acute pancreatitis and they have higher mortality rate in comparison to those having SLE only14.
The incidence of MAS is also high in ASOD that closely resembles sJIA. It is reported to be 7.7-16% with the mortality rate ranging between 9.5-22%15. The incidence of MAS in SLE is about 0.9-4.6%16.
Features of MAS
The clinical features of MAS evolve rapidly. The common signs and symptoms presented by the patients are non-remitting high fever, hepatomegaly, splenomegaly. Neuro-psychiatric dysfunction, lymphadenopathy, pancytopenia, skin erythema, anemia and less frequent abnormalities of heart, kidneys and lung tissues17,7,18 (Table 1).
Table (1):
Signs and symptoms of MAS [20]
Features |
Incidence |
|---|---|
Fever |
95% |
Arthritis |
64% |
Hemorrhagic abnormalities |
20% |
Lymphadenopathy |
51% |
Hepatomegaly |
70% |
Splenomegaly |
55% |
Neurologic dysfunction |
34% |
Involvement of tissues (heart, lung, kidney) |
10% |
The laboratory results in MAS are not specific, however the frequently observed changes (Table 2) are increase in hepatic enzymes, bilirubin, triglycerides, ferritin, D-dimer and lactate dehydrogenase while there is decrease in levels of sodium, hemoglobin, blood cells like leucocytes and platelets, albumin, fibrinogen, clotting factors like II, VII and X. Patients also show elevated partial thrombo-plastin time along with the presence of degradation product of fibrinogen. Levels of various cytokines are also found to be elevated. Some of the examples are interleukins (IL-1, IL-2, IL-6), tumor necrosis factor a (TNF a), interferons (INF a, INF g) and macrophage colony stimulating factor (MCSF)17.
Table (2):
Laboratory changes in MAS
Investigations |
Changes |
|---|---|
1. Hematological profile
Hemoglobin White blood cell count Platelet count INR and PTT ESR |
Decreases Decreases Decreases Increases Decreases |
2. Biochemical profile
Ferritin D-dimer Liver enzymes LDH TG SCD 25 (receptor for IL-2) and SCD 163 (receptor for hemoglobin-haptoglobin complex) Fibrinogen |
Increases Increases Increases Increases Increases Increases
Decreases |
3. Histopathological changes
Bone marrow biopsy |
Increased hemophagocytic macrophages with elevations in CD163 |
Bone marrow biopsy shows the highly elevated number of well differentiated macrophages that actively phagocytize hemato-poietic cells. However hemophagocytosis may not be detectable during the initial stages and may lack sensitivity in 40% of cases19. Other tissues like lymph node, spleen and liver also exhibit hemophagocytic pattern but these sites are rarely explored in literatures.
Type of MAS
MAS is basically categorized as subtype of secondary hemophagocytic-lymphohisto-cytosis (sHLH)21. HLH is broadly categorized into two distinct types as:
Primary HLH ( inherited or familial form)
Secondary HLH ( acquired form)19,22.
Primary HLH is autosomal recessive in nature with high incidence in infants (80% cases) compared to adults23. It is characterized by the following features:
Defective T-cell activation
Defective NK cell mediated cytolytic pathway
Excess production of cytokines that result in uncontrolled activation T-cells and macrophages24.
Secondary HLH can occur at any ages due to excess immunological activation as a result of various infections caused by viruses (Epstein barr, Herpes simplex, Varicella-zoster), bacteria (Hemophilus, Mycobacterim, Brucela), fungus (candida, Histoplama), parasites (Leishmania), drugs, hematological malignancies and autoimmune inflammatory disorders22,25.
Diagnosis
It is often difficult and challenging to properly diagnose MAS as it can mimic infections or malignancies or may be present as the complications of sJIA. Diagnosis is also difficult in ealrly phases due to lack of pathognomic clinical findings. In 1991, the criteria for diagnosis of MAS was first proposed by a scientific team studying histocyte disorders. These criterias were later modified in 2004 25. According to this modified diagnostic guideline, the patient is supposed to be suffering from MAS if any one of the following characteristics is met:
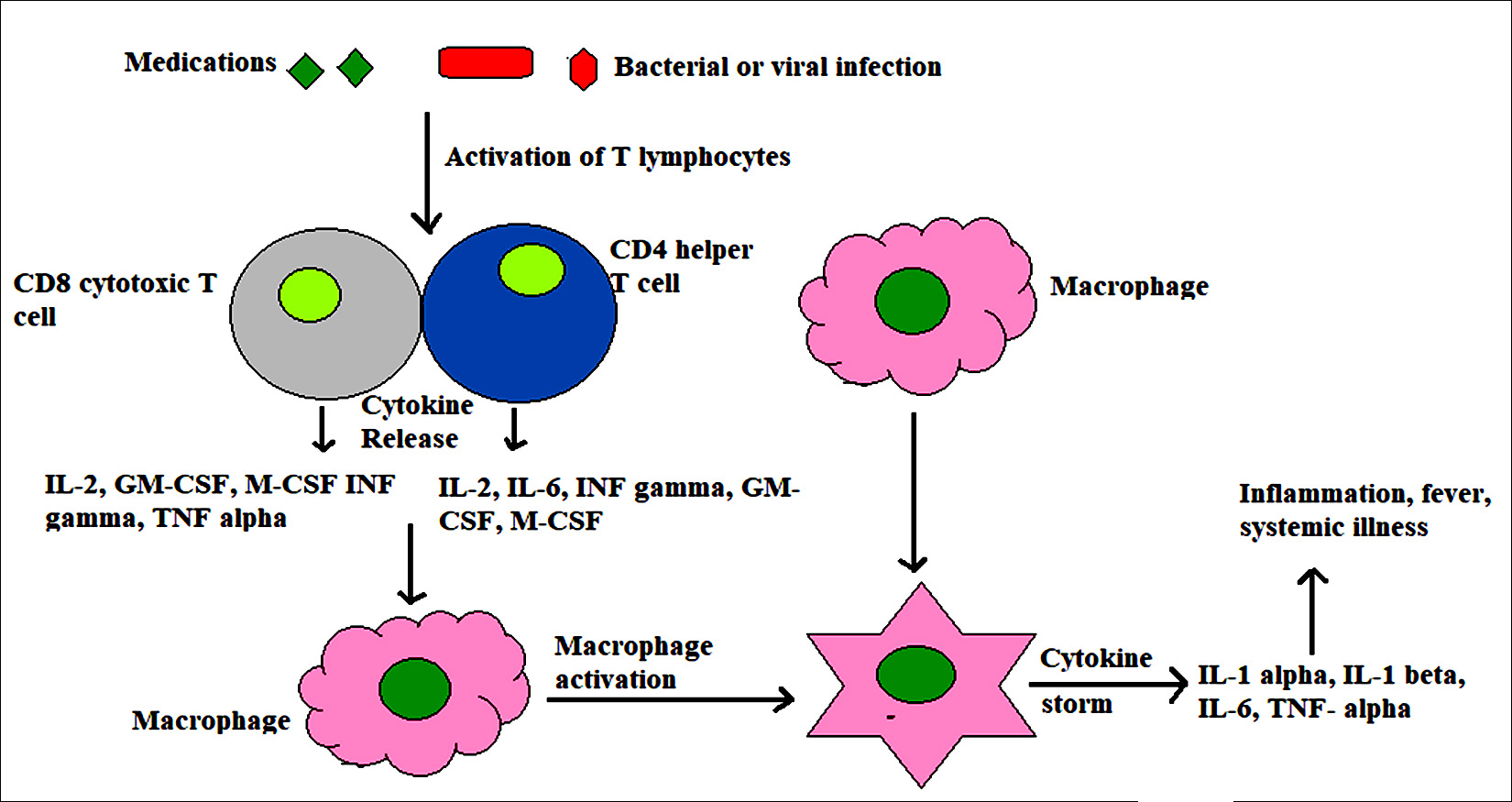
Fig. 1. Macrophage activation syndrome induced by infections and medications. Under the stimulation of bacterial, viral or fungal infection and certain medicines, T cells are activated and may directly induce apoptosis of both activated macrophages and/or T cells themselves. It results in dysregylation of cytotoxic activities causing persistent of macrophages and T cells leading to cytokine storm.
Presence of gene mutations (PRF, UNC13D, STX11) associated with HLH18 or
5 out of 8 laboratory and clinical diagnostic criterias. These include cytopenia, increased levels of triglyceride/fibrinogen, hemophagocytosis, low or absence of NK cell activity. Increase in ferritin and soluble CD25, fever and hepatospleenomegaly21 (Table 3).
Table (3):
Diagnostic criteria of MAS as formulated in 2004 [18]
| Criterias | |
|---|---|
| 1. Molecular diagnosis having specific gene mutation consistent with HLH | |
| 2. Clinical and laboratory criteria (5 out of 8 are to be met→ 2 clinical and 3 laboratory) | |
| A. Clinical
i. Fever ii. Splenomegaly |
|
| B. Laboratory
i. Cytopenia a. Hemoglobin b. Platelets c. Neutrophils ii.Hypertriglyceridemia and/or hypofibrinogenemia iii. Hemophagocytosis iv. Decreased or absence of NK cell activity v. Increased ferritin vi. Increased soluble CD25 (Il-2 receptor) |
<9 gm/dl (adult), <10 gm/dl (infant<4 weeks) <10 x 109/dl <1 x 108/dl >265 mg/dl <0.15gm.dl Bone marrow aspirate, spleen or lymph nodes – >500 mg/dl >2400U/ml |
However, several researches have focused on challenges in applying 2004 diagnostic criteria for MAS, as sJIA, the most frequently associated rheumatologic disorder with MAS26 is independently associated with hyperferritinemia, anemia27 and leucocytosis28 in absence of MAS. Such overlap of clinical features may pose difficulty in early diagnosis of MAS onset29. Likewise coagulopathies which are not only common in MAS30 but also in other disorders can further complicate the sample collection procedure for histological diagnosis25. To address these issues, Raveli et al.7 proposed a preliminary diagnostic criteria for MAS associated with sJIA in 2005. According to this guideline, diagnosis of MAS needs the fulfillment of ³2 laboratory and ³2 clinical criterias (Table 4).
Table (4):
Diagnostic criteria for MAS complicating sJIA [7]
Criteria |
Changes |
|---|---|
A. Clinical
1. Neurologic dysfunction
2. Hemorrhages 3. Hepatomegaly |
Headache, lethargy, irritability, disorientation, seizures and come Purpura, ecchymoses and mucosal bleeding Liver enlargement≥3cm below coastal margin |
B. Laboratory
Decreases platelet count Decreased leucocyte count Decreased fibrinogen level Increased AST levels |
≤262 x 106/ µl ≤4 x 106/ µl
>59 U/L |
The preliminary diagnostic guideline of Raveli et al were assessed by Davi et al, who compared it with that of 2004 diagnostic guidelines. They concluded that preliminary diagnostic criteria showed best results with high sensitivity and specificity of 86%31. Another diagnostic criteria for MAS associated with sJIA as also approved by European league agains Rheumatism/ American College of Rhematology (EULAR/ACR) in 2016 (Table 5)32,33.
Table (4):
EULAR/ACR criteria for MAS in sJIA
| Patients with sJIA having | |
|---|---|
| 1. a. Fever
b. Increased ferritin |
>684 ng/ml |
| 2. And any two of the following criterias
i. Platelet count ii. AST level iii. Triglyceride level iv. Fibrinogen level |
<181 x 109/L >48 U/L >156 mg/dl <360 mg/dl |
Cytokine storm in MAS
Many studies have reported high levels of cytokines, TNF receptors and IL-1R antagonists in MAS34. Cytokines present include proinflam-matory cytokines like those derived from lymphocytes (INF g, IL-2) and cytokines produced by monocytes and macrophages (IL-1b, TNF g, IL-6 and IL-18). Regulatory cytokines such as IL-10 are also elevated in MAS34. Patients with severe MAS may disturb the regulatory pathways of IL-10 which can lead to uncontrolled inflammation. Stimulation of toll like receptor 9 along with blockade of IL-10 receptor can lead to more severe complications35.
IL-1a, a proinflammatory cytokine, is considered to be the central one in pathogenesis of disease. It is initially secreted in an inactive form but when cells are activated, caspase-1 protein cleaves pro IL1a to its active form. On activation, it causes further activation of lymphocytes and endothelial cells thereby stimulating production of other inflammatory cytokines like IL-636.
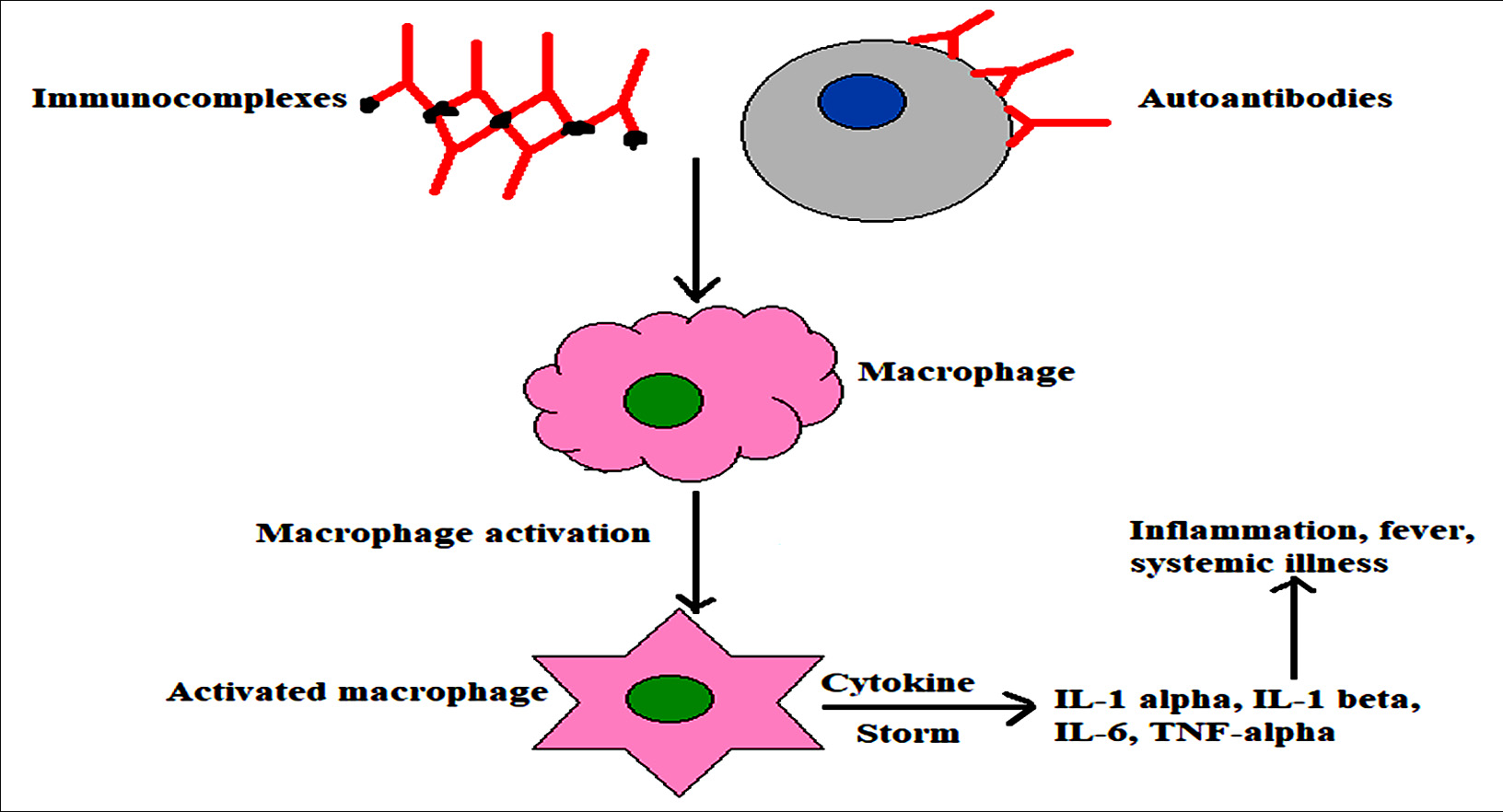
Fig. 2. Auto-antibodies or immune complexes can also stimulate macrophages resulting in production of enormous amount of cytokines
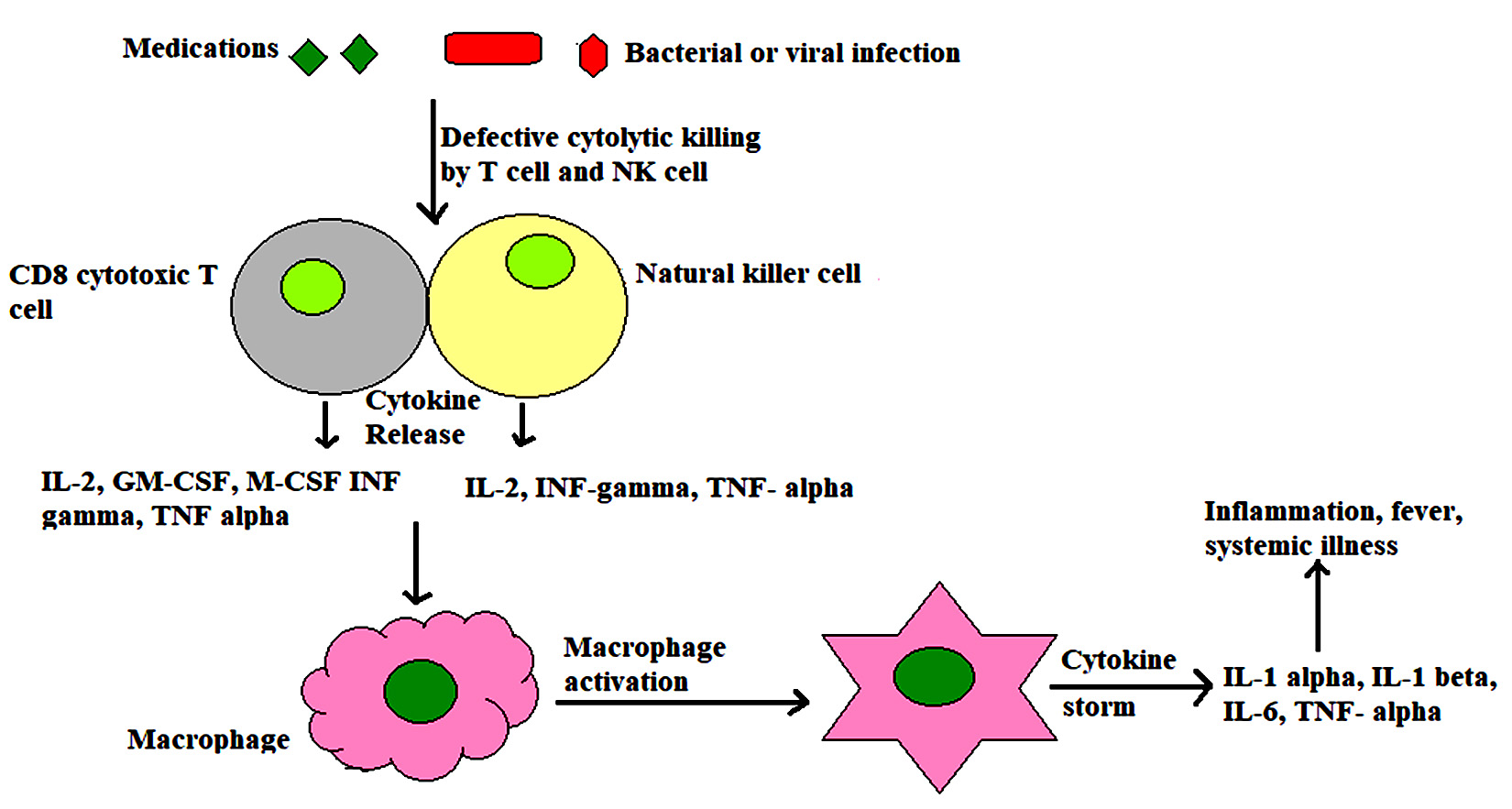
Fig. 3. Failure of cytolytic killing mechanism of T cells and NK cells due some genetic defects (such as defect in perforin gene), can also promote MAS, as defective cytotoxicity casuses continuum of antigenic stimulation leading to persistent T and NK cell activation and proliferation. Cytokines are persistently released from activated cells that further stimulate macrophages leading to cytokine storm.
Pathophysiology
The etiology and specific pathogenesis of MAS are yet to be clearly elucidated. However most of the theories derived rely on the study of primary HLH which is clinically quite similar to MAS. It is stated that hemophagocytosis can develop due to dysregulation of immunologic phenomenons resulting in the exaggeration of inflammatory responses which is characterized by excessive activation of macrophages and T lymphocytes that ultimately leads to cytokine storm37.
It has been reported that numerous proinflammatory cytokines are released. It is also demonstrated that hyperactive T- cells and macrophages reside in various organs and produce cytokines like INF g, TNF a, IL-6 and IL-1 which play a major role in pathogenesis of MAS. The condition is further aggravated by the presence of perforin deficiency38. Perforin is a type of cytotoxic protein secreted by T- cells and NK cells to destroy virus infected cells. It is also involved in regulation of lymphocyte prolife-ration. Mutation in gene coding for perforin is linked with MAS as it can lead to T-cell and NK cell dysfunction39,40 and persistent lymphocyte activation38.
IL-1b via its receptors signals production of IL-641 that plays a central role in inducing acute phase response40. Some researchers have shown that imbalance between IL-18 and its inhibitor results in activation of macrophages and Th-1 lymphocytes that have bypassed NK cell mediated cytotoxic control, leading to the manifestation of sHLH43-42. IL-18 belongs to IL-1 family and stimulates the production of INF g (by NK cells and T-cells), TNF g and chemokines (by macrophages)41. The major role of INF g is to cause profound activation of macrophages and monocytes. The increased levels of INF g is also well correlated with clinical and laboratory features of MAS43.
Table (6):
Major cytokines inducing MAS
Cytokines |
Roles |
|---|---|
IL-1α and IL-1β |
Co-stimulation of T cell and antigen presenting cell
Acute phase response Fever and inflammation Hematopoeisis |
IL-2 |
Proliferation and activation of B cells, T cells and NK cells |
IL-6 |
Proliferation pf B cell
Acute phase response Stimulation of T cells synergistically with TNF-α and IL-1 |
INF-γ |
Promotes cell mediated immunity
Activates neutrophils, NK cells and macrophages |
TNF-α |
Activates signaling pathways that cause proliferation of immune cells |
GM-CSF and M-CSF |
Stimulate proliferation and differentiation of dendritic cells and monocytes |
Treatment
There is necessity of urgent management of MAS. The preliminary purpose of the therapy used should be control of hyper inflammatory state with the use of immunosuppressive or cytotoxic drugs. The most commonly used drugs are corticosteroids and cyclosporins. However other therapeutic agents like etanercept, etoposide, immunoglobins in high dose and plasmapheresis are also used in practice44.
Steroids such as methylprednisolone and dexamethasone are the first line drugs of choice as the response of patients is usually prompt. However steroids are rarely used as monotherapy for long term due to their side effects45. In case of MAS unresponsive to steroids alone, a combination therapy consisting of steroids and cyclosporine is given. Cyclosporin is an inhibitor of T-cell function and mostly administered in early stage of the syndrome25. It also has membrane stabilizing effect on macrophages46.
Etoposide is a bone marrow suppressant and its use can result in severe infection, therefore caution is to be taken in patients having renal or hepatic impairment47. As per Coca et al, antithymocyte globulin (ATG) could be used in place of etoposide48 but it may be associated with infusion reactions49.
In case of MAS associated with sJIA, IL-1 inhibitor (such as anakinra) is being used increasingly50. Though the exact role of IL-1 in pathogenesis of MAS is unclear, it is expected that IL-1 inhibition may aid in better control of the disease. According to the several reports, Il-1 inhibitors have proven better treatment choice in MAS complicating sJIA, which showed inadequate response to cyclosporins and corticosteroids51.
Successful treatment of MAS associated with ASOD using IL-1 blockers have also been documented46. However according to the recent report, of 23 patients with sJIA who were treated with anakinra, one patient developed MAS52 and in another report it was stated that anakinra might have triggered MAS in 5 out of 46 patients of sJIA53. IL-6 blockers (like tocilizumab) is also used as the treatment strategy among the patients who could not tolerate IL-146.
Activated macrophages produce IL-6 which may further amplify the macrophage response to the proinflammatory stimuli54. However, as per the Japaneses study, tocilizumab when used in the treatment of ASOD gradualy progressed the patient to MAS55. Reports regarding development of cytopenias following the use of tocilizumab in patients with sJIA (complicated with MAS) are also available56. Rituximab and intravenous Igs have shown satisfactory results in MAS induced by viruses like Epstein barr virus and cytomegalo virus57.
MAS, though rare, is a potentially fatal disorder that requires prompt and appropriate treatment to prevent deleterious outcomes. Since there is lack of valid criteria for the diagnosis of MAS, the treatment appears highly variable at different clinical sectors. However the most commonly used treatment is the use of corticosteroids as monotherapy or along with cyclosporins or immunoglobulins. In some cases use of etoposide and IL-1 bockers are also recommended.
As MAS is mostly associated with rheumatic disease, clinicians should be aware of diagnostic principle and management. It is necessary to understand the underlying pathophysiology of MAS in order to identify the pathways involved in early phase of the syndrome so that better ideas could be developed in furnishing newer treatment biomarkers. Additionally larger cohorts regarding immuno-logical and genetic studies must be carried out to formulate more effective and appropriate therapy as well as to improve mortality. Also studies have shown that the cytokine storm is the main driving force for pathology of hyper-inflammatory syndromes. Therefore therapeutic approaches blocking cytokine function might be fruitful with less toxic effects.
None
The authors declare that there is no conflicts of interest.
- Young L, Yap L, and Murray P. Epstein-Barr virus: more than 50 years old and still providing surprises. Nature Reviews: Cancer, 2016; 16(12): 789–802.
- Jha H, Pei Y, and Robertson E. Epstein-Barr virus: diseases linked to infection and transformation. Frontiers in Microbiology, 2016; 7: 1602.
- Shinozaki A, Kunita A, and Fukayama M. Update on Epstein-Barr virus and gastric cancer (review). International Journal of Oncology, 2015; 46(4): 1421–1434.
- Jang B, Jung E, and Kim W. Expression of Bam HI-A rightward transcripts in Epstein-Barr virus-associated gastric cancers. Cancer Research and Treatment, 2011; 43(4): 250–254.
- Murata T, Noda C, and Narita Y. Induction of Epstein-Barr virus oncoprotein LMP1 by transcription factors AP-2 and early B cell factor. Journal of Virology, 2016; 90(8): 3873–3889.
- Decaussin G, Lammali F, Turenne M, Bouguermouh A, and Ooka T. Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Research, 2000; 60(19): 5584–5588.
- Zhang C, Decaussin G, Daillie J, and Ooka T. Altered expression of two Epstein-Barr virus early genes localized in Bam HI-A in non-producer Raji cells. Journal of Virology, 1988; 62(6): 1862–1869.
- Takada K. Role of EBER and BARF1 in nasopharyngeal carcinoma (NPC) tum-origenesis. Seminars in Cancer Biology, 2012; 22(2): 162–165.
- Hoebe E, Wille C, E. and Hopmans E. Epstein-Barr virus transcription activator R upregulates BARF1 expression by direct binding to its promoter, independent of methylation. Journal of Virology, 2012; 86(20): 11322–11332.
- Liu M, Shih Y, and Li L. Expression of the Epstein-Barr virus BHRF1 gene, a homologue of Bcl-2, in nasopharyngeal carcinoma tissue. Journal of Medical Virology, 2000; 61(2): 241–250.
- Luo B, Wang Y, and Wang X. Expression of Epstein-Barr virus genes in EBV-associated gastric carcinomas. World Journal of Gastroenterology, 2005; 5: 629–633.
- Rameshkumar K, Clark D J. Efficient extraction of DNA from paraffin- embedded tissue using nucleon HT kit for clinical molecular pathology research. Life Science News: Biosciences: 1998.
- Telenti A, Marshall W , Smith T . Detection of Epstein Barr Virus by polymerase chain reaction. Journal of clinical microbiology, 1990; 28(10): 2187-2190.
- Hashimoto K, Shimizu Y, Suehiro Y. Hypermethylation status of APC inversely correlates with the presence of sub-mucosal invasion in laterally spreading colorectal tumors. Mol Carcinogen 2008; 47: 1–8.
- Jing L, Wen L, Kui C. The methylation status and expression of Epstein Barr Virus early genes BARF1 and BHRF1 in Epstein Barr Virus associated gastric carcinomas. Hindawi gastroenterology research and practice. 2017, Article ID 3804146.
- Jones P, Baylin S. The fundamental role of epigenetic events in cancer. Nat Rev Genet. 2002; 3(6): 415–28.
- Fukayama M, Ushiku T. Epstein-Barr virus-associated gastric carcinoma. Pathol Res Pract. 2011; 207(9):529–37.
- Sheng W, Decaussin G, Ligout A, Takada K, and Ooka T. Malignant transformation of Epstein-Barr virus-negative Akata cells by introduction of the BARF1 gene carried by Epstein-Barr virus. Journal of Virology, 2003; 77(6): 3859–3865.
- Decaussin G, Sbih F, Turenne M, Bouguermouh A, and Ooka T. Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Research, 2000; 60(19): 5584–5588.
- Hoebe E, Le Large E, Greijer A, and Middeldorp J. BamHI-A rightward frame 1, an Epstein-Barr virus-encoded oncogene and immune modulator. Reviews in Medical Virology, 2013; 23(6): 367–383.
- Hoebe E, Wille C, and Hopmans E. Epstein-Barr virus transcription activator R upregulates BARF1 expression by direct binding to its promoter, independent of methylation. Journal of Virology, 2012; 86(20): 11322–11332.
- Dawson C, Eliopoulos A, Dawson J, and Young L. BHRF1, a viral homologue of the Bcl-2 oncogene, disturbs epithelial cell differentiation. Oncogene, 1995; 10(1): 69–77.
© The Author(s) 2019. Open Access. This article is distributed under the terms of the Creative Commons Attribution 4.0 International License which permits unrestricted use, sharing, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.